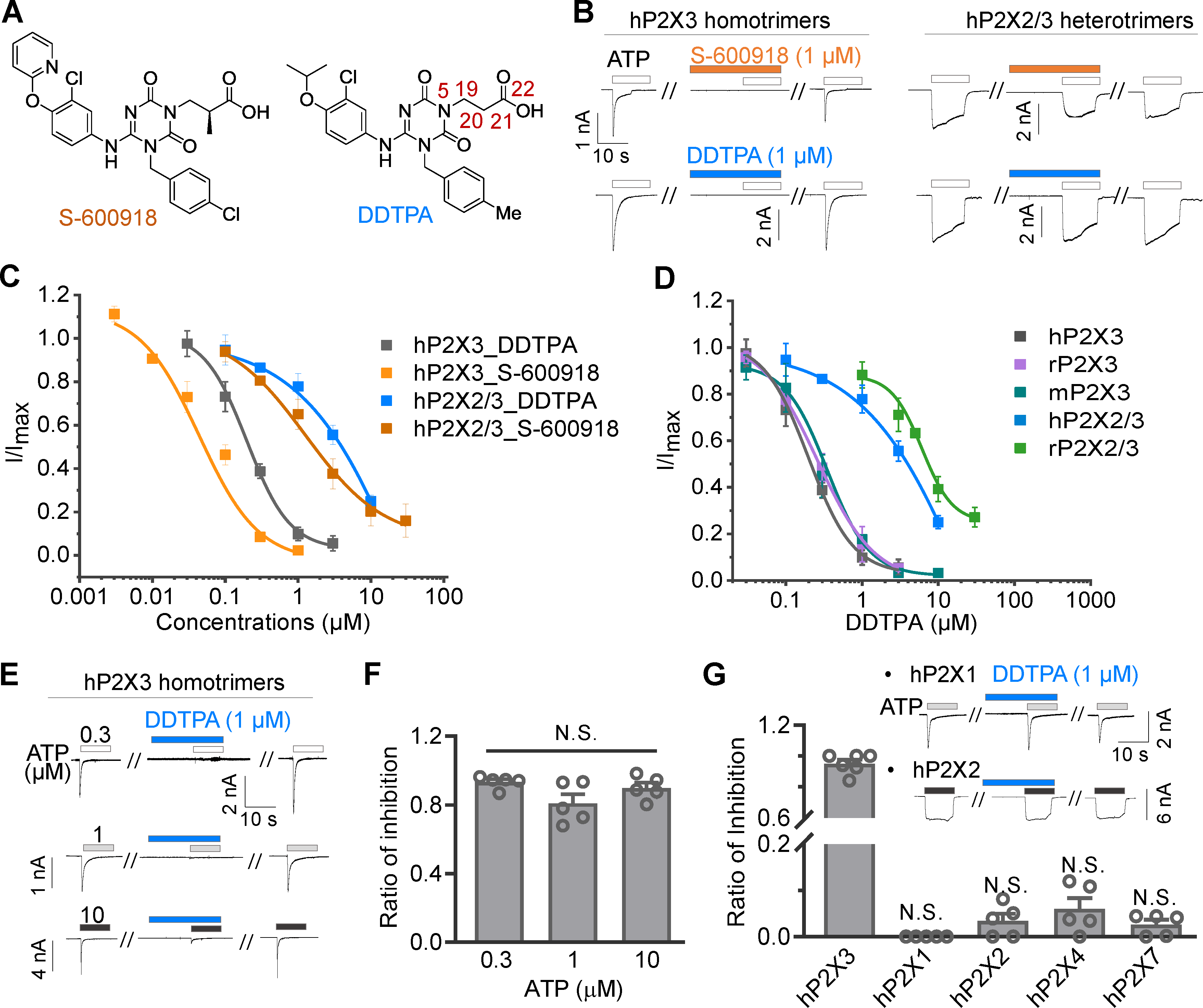
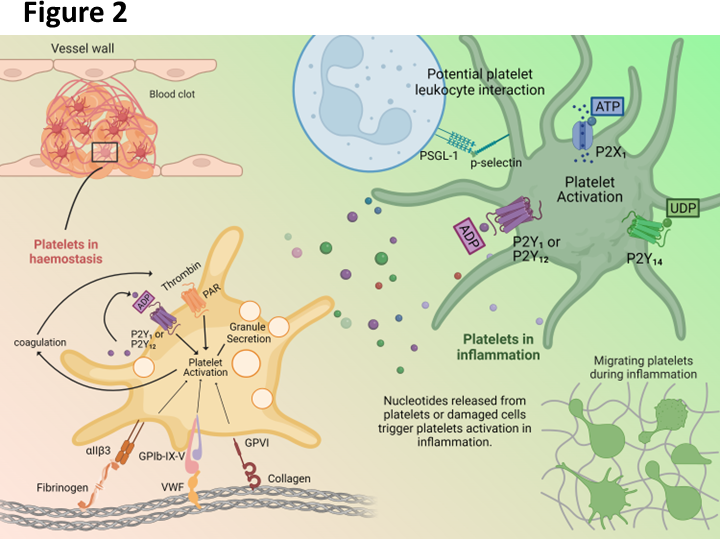
Background and Purpose: The ionotropic purinergic trimeric receptor P2X3 is a new drug target other than the opioid receptor for the treatment of refractory chronic cough (RCC). However, the only marketed P2X3 antagonist, Gefapixant/AF-219, has a side effect of taste disorders due to simultaneous action on the human P2X2/3 (hP2X2/3) heterotrimer. Therefore, selective molecules with high affinity for the hP2X3 homotrimer and low affinity for the hP2X2/3 heterotrimer have potential in iteration 2.0 RCC drug development, such as Sivopixant/S-600918, a clinical phase II RCC candidate with lower taste disturbance than Gefapixant. S-600918 and its analogue (3-(4-((3-chloro-4-isopropoxyphenyl)amino)-3-(4-methylbenzyl)-2,6-dioxo-3,6-dihydro-1,3,5-triazin-1(2H)-yl)propanoic acid (DDTPA) exhibit both high affinity and high selectivity for hP2X3 homotrimers compared to hP2X2/3 heterotrimer. The mechanism of its druggable site and this high selectivity is not clear. Experimental Approach: Here, we reveal a novel allosteric mechanism that distinguishes this drug candidate from other P2X3 inhibitors through chimera construction, site covalent occupation, metadynamics, mutagenesis, and electrophysiology. Key Results: We suggest that the tri-symmetric site adjacent to the upper vestibule determines the high affinity and selectivity of S-600918/DDTPA for hP2X3. Only four amino acids of the hP2X2 upper body domain swapped with hP2X3, allow the hP2X2/3 heterotrimer to gain comparable affinity for S-600918/DDTPA as the hP2X3 homotrimer. Conclusion and Implications: Thus, we have revealed the molecular basis for the cough suppressive effects and reduced side effects of new RCC clinical candidates from the perspective of receptor-ligand recognition, which may provide information critical for the development of new drugs targeting P2X3 for indications such as RCC, idiopathic pulmonary fibrosis (IPF), and primary hypertension.