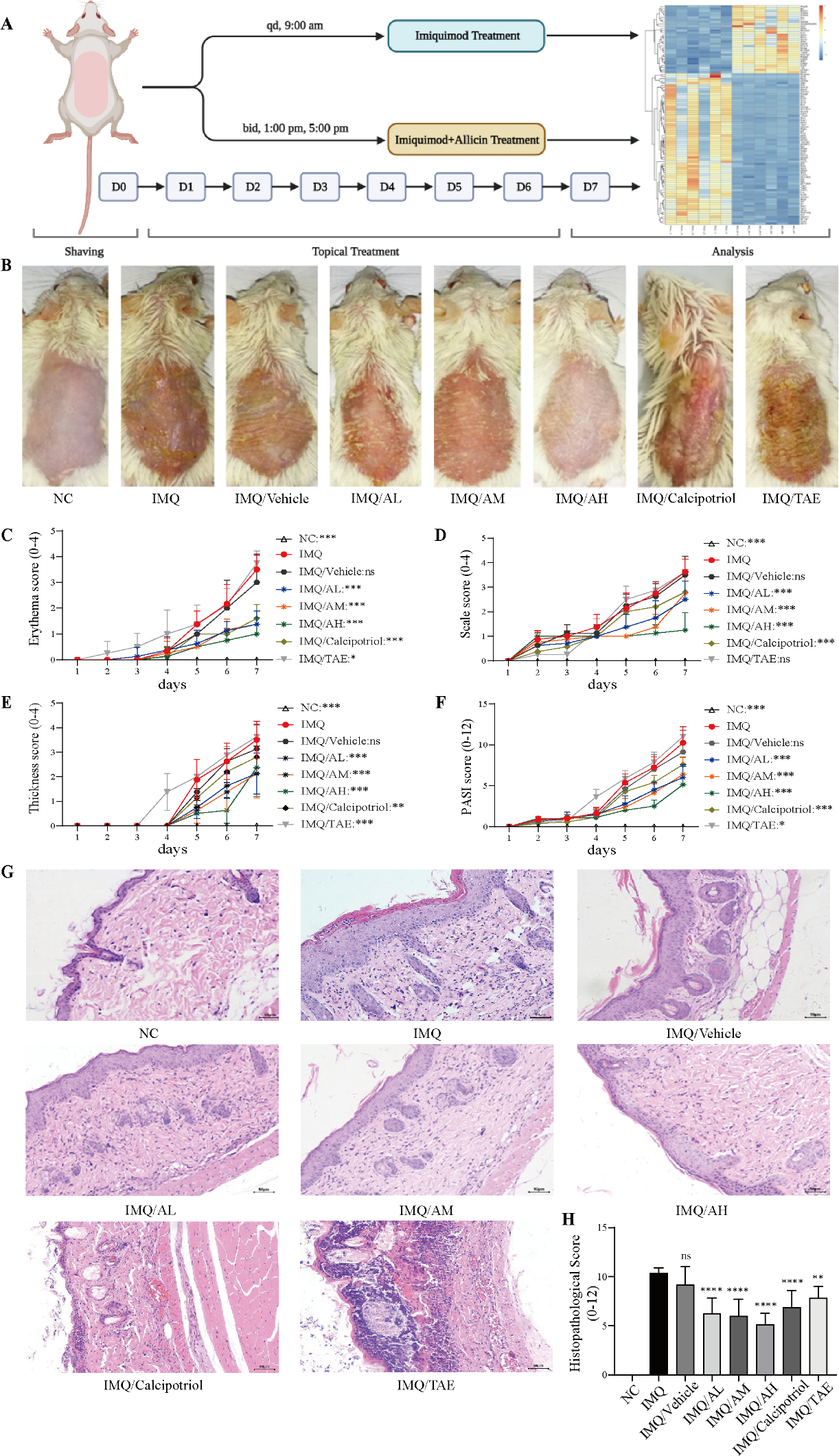
Background and Purpose: Psoriasis is an inflammatory skin disease of chronic recurrence mediated by the interaction between IL-17 and keratinocytes, which sustains a vicious circle of inflammation. Currently, there is no safe and effective natural medicine for the clinical treatment of psoriasis. Given its prominent anti-proliferative and anti-inflammatory properties, we investigated the mechanism of allicin improving psoriasis. Experimental Design: Pharmacodynamics and toxicology experimental studies were estimated after topical administration of allicin on the skin of mice. Changes in inflammatory factors expression were analyzed by qPCR and immunohistochemistry after topical treatment with allicin in mice with psoriasis-like lesions induced by imiquimod. The impacts of allicin on proliferation and apoptosis of keratinocytes were analyzed by CCK8 assay and flow cytometry. The interaction between IL-17A and keratinocytes was studied using HaCaT cells, and the mechanism of action of allicin was explored by Western Blot. Transcriptomic changes following the action of allicin were probed by RNA-seq. Key Results: Our study demonstrated that allicin significantly improved the epidermal structure by inhibiting excessive proliferation and evasion of apoptosis of keratinocytes. Furthermore, allicin reduced the secretion of inflammatory cytokines (IL-17A/F, IL-22, IL-12, IL-20), chemokines (CXCL2, CXCL5, CCL20), and antibacterial peptides (S100A8/9). Mechanistically, allicin directly inhibited the IL-17-induced TRAF6/MAPK/NF-κB and STAT3/NF-κB signaling cascades in keratinocytes, thus breaking the positive inflammation feedback and alleviating imiquimod-induced psoriasis-like dermatitis in mice. Importantly, topical administration of allicin did not cause skin allergy, and the safety and adaptability of long-term application were verified.
/FIbrosis mechanisms (7).png?1648618865)