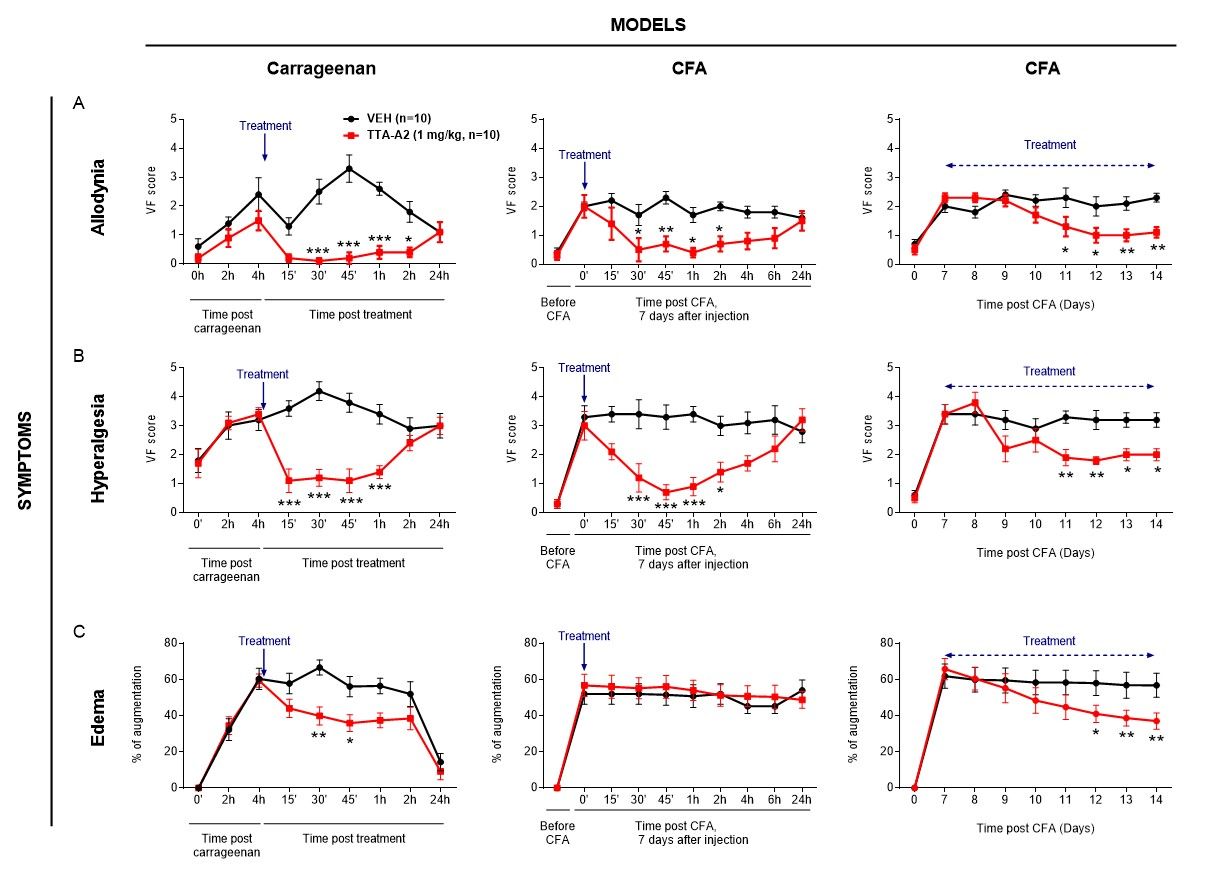
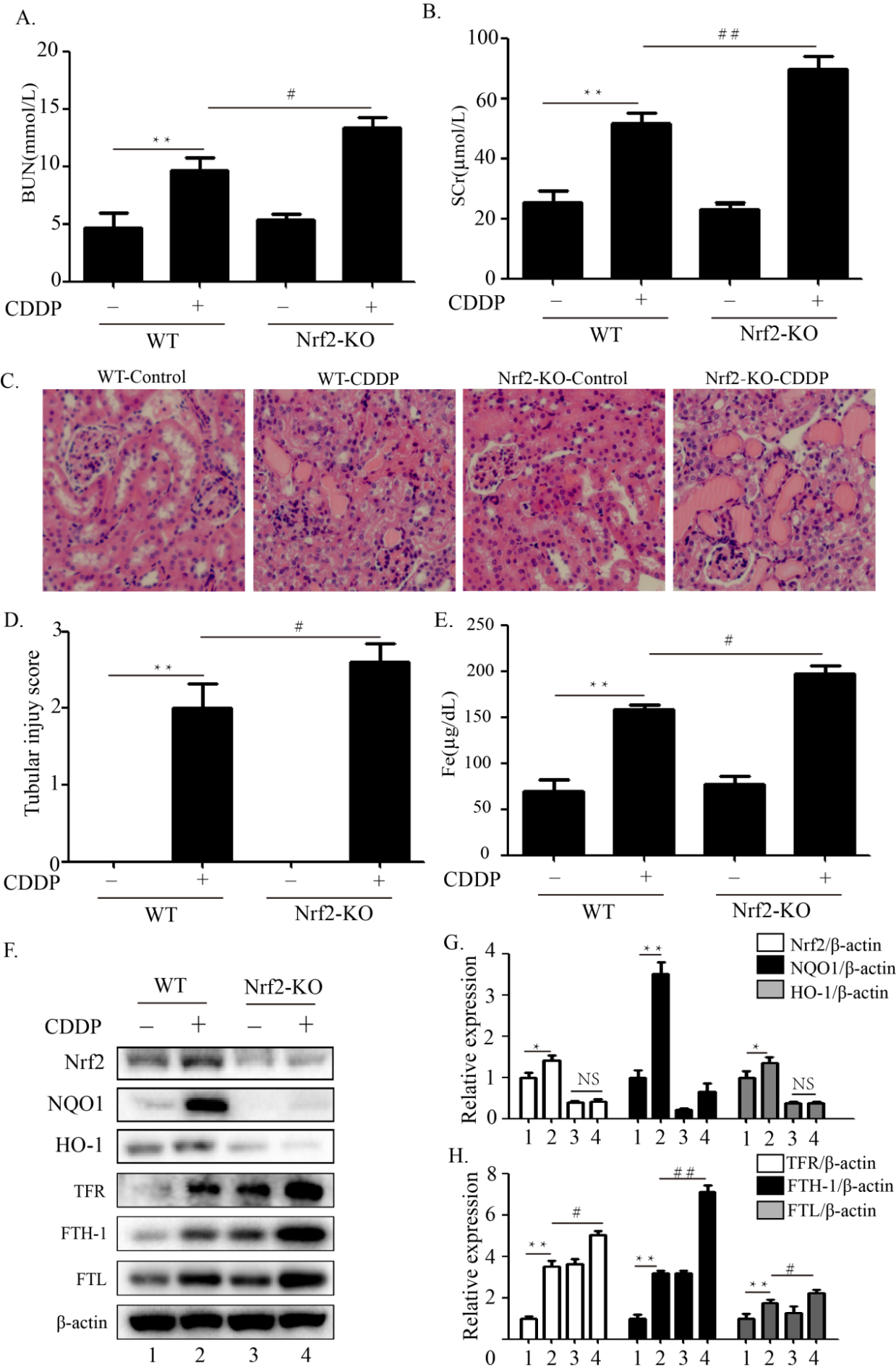
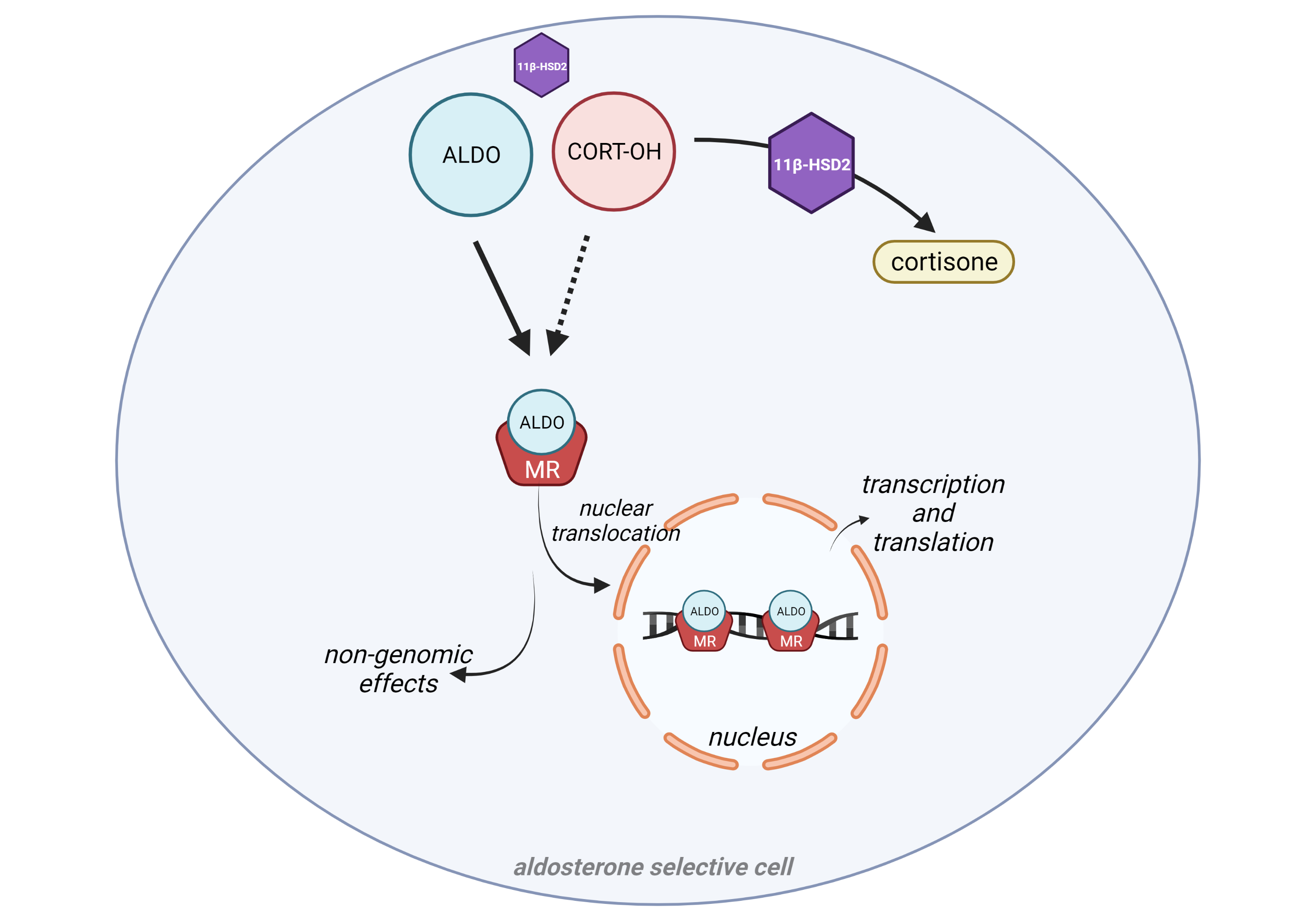
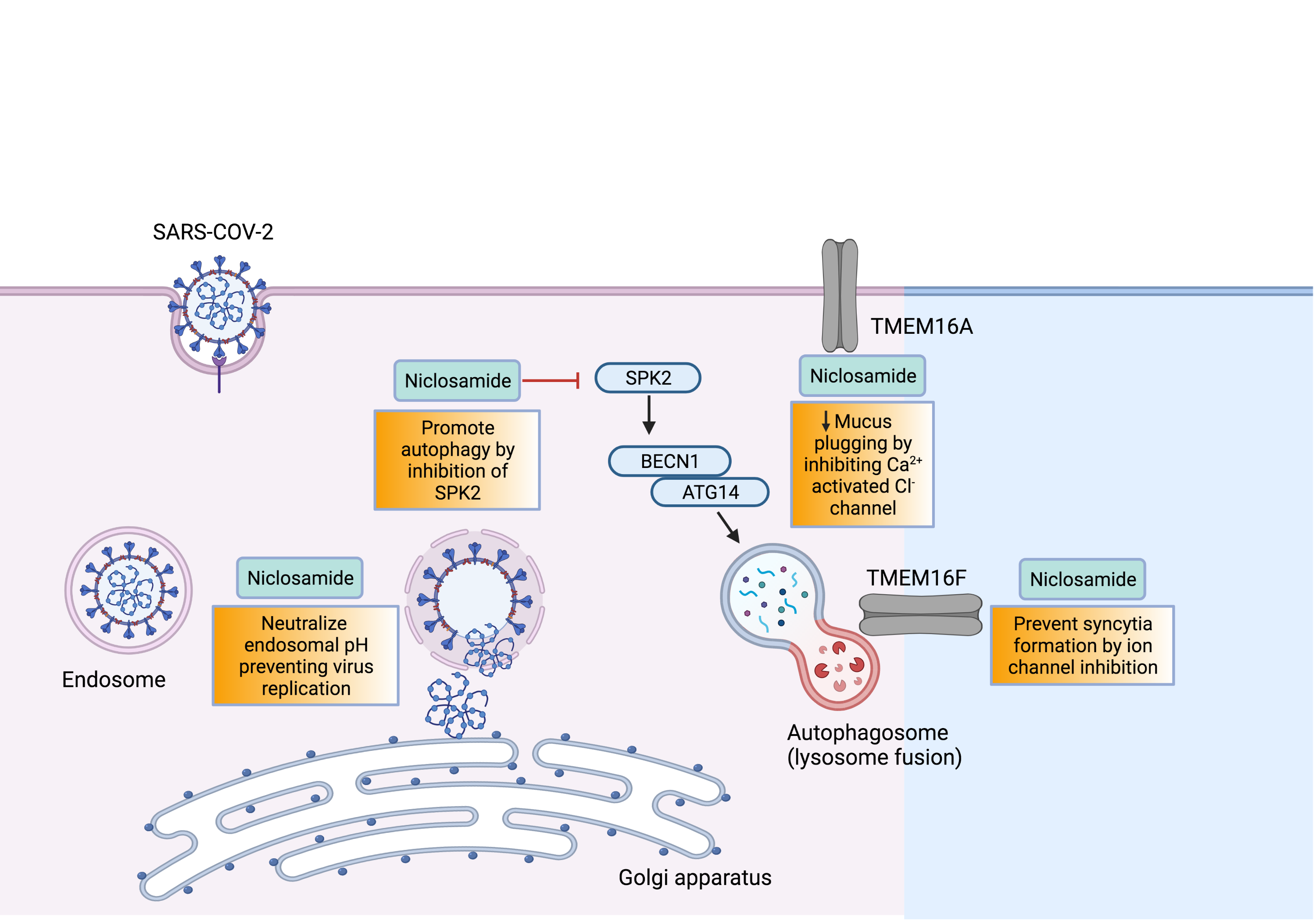
Background and Purpose. The proalgesic transient receptor potential (TRP) ankyrin 1 (TRPA1) channel, expressed by a subpopulation of primary sensory neurons, has been implicated in various pain models in mice. However, evidence in rats indicates that TRPA1 conveys nociceptive signals elicited by channel agonists but not those associated with tissue inflammation or nerve injury. Here, in rats, we explored the TRPA1 role in mechanical allodynia associated with neurogenic inflammation and moderate (partial sciatic nerve ligation, pSNL) or severe (chronic constriction injury, CCI) sciatic nerve injury. Experimental Approach. Acute nociception and mechanical hypersensitivity associated with neurogenic inflammation and sciatic nerve injury (pSNL and CCI) were investigated in rats with TRPA1 pharmacological antagonism or genetic silencing. TRPA1 presence and function was analyzed in cultured rat Schwann cells. Key Results. Hind paw mechanical allodynia (HPMA), but not acute nociception, evoked by local injection of the TRP vanilloid 1 (TRPV1) agonist, capsaicin, or the TRPA1 agonist, allyl isothiocyanate, was mediated by calcitonin gene related peptide (CGRP) released from peripheral nerve terminals. CGRP-evoked HPMA was sustained by a reactive oxygen species (ROS)-dependent TRPA1 activation, probably in Schwann cells. HPMA evoked by pSNL, but not that evoked by CCI, was mediated by ROS and TRPA1 without the involvement of CGRP. Conclusions and Implications. As found in mice, TRPA1 mediates mechanical allodynia associated with neurogenic inflammation and moderate nerve injury in rats. The channel implication in mechanical hypersensitivity following inflammation and partial nerve damage is a common rodent feature and might be explored in humans.
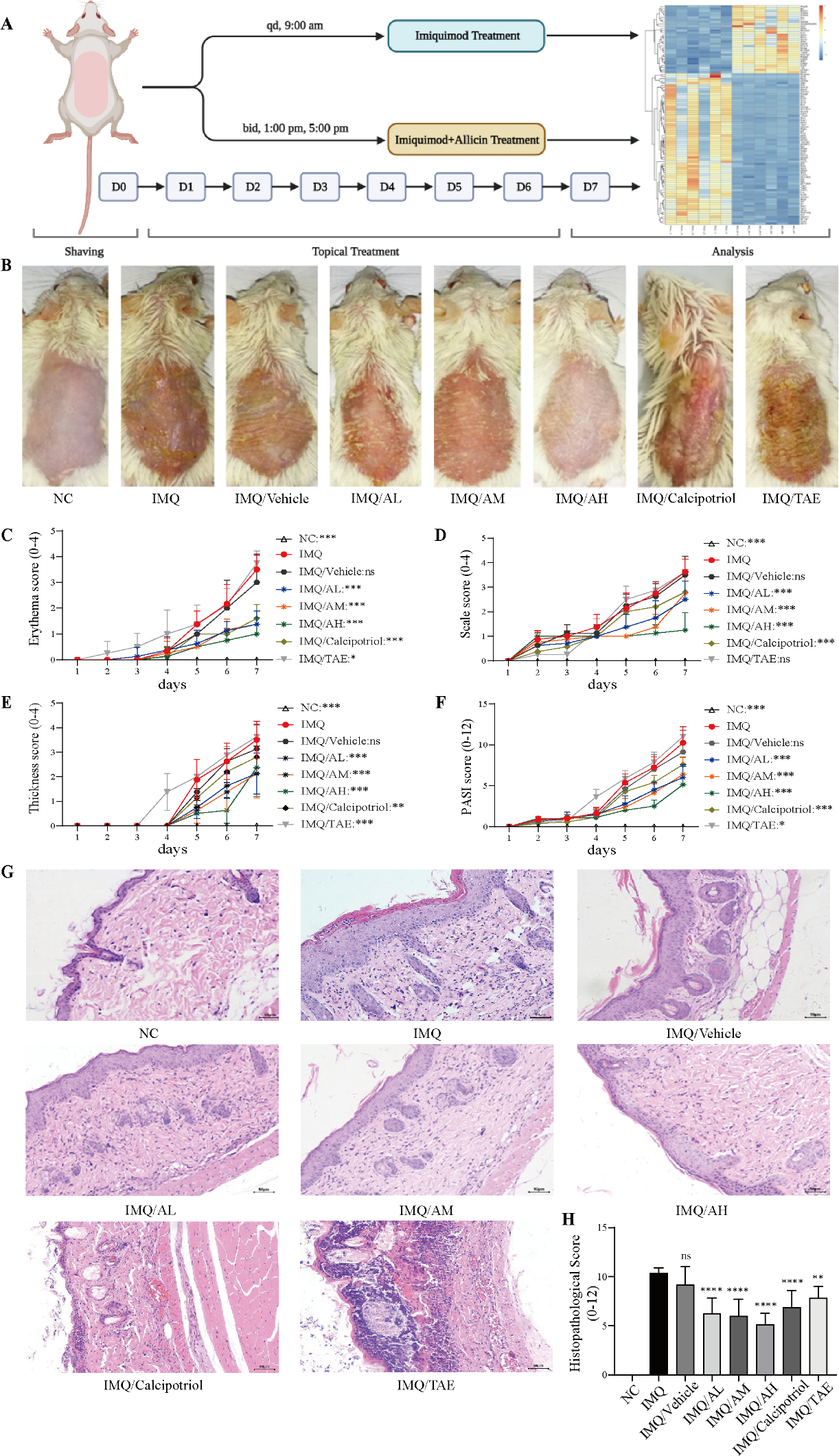
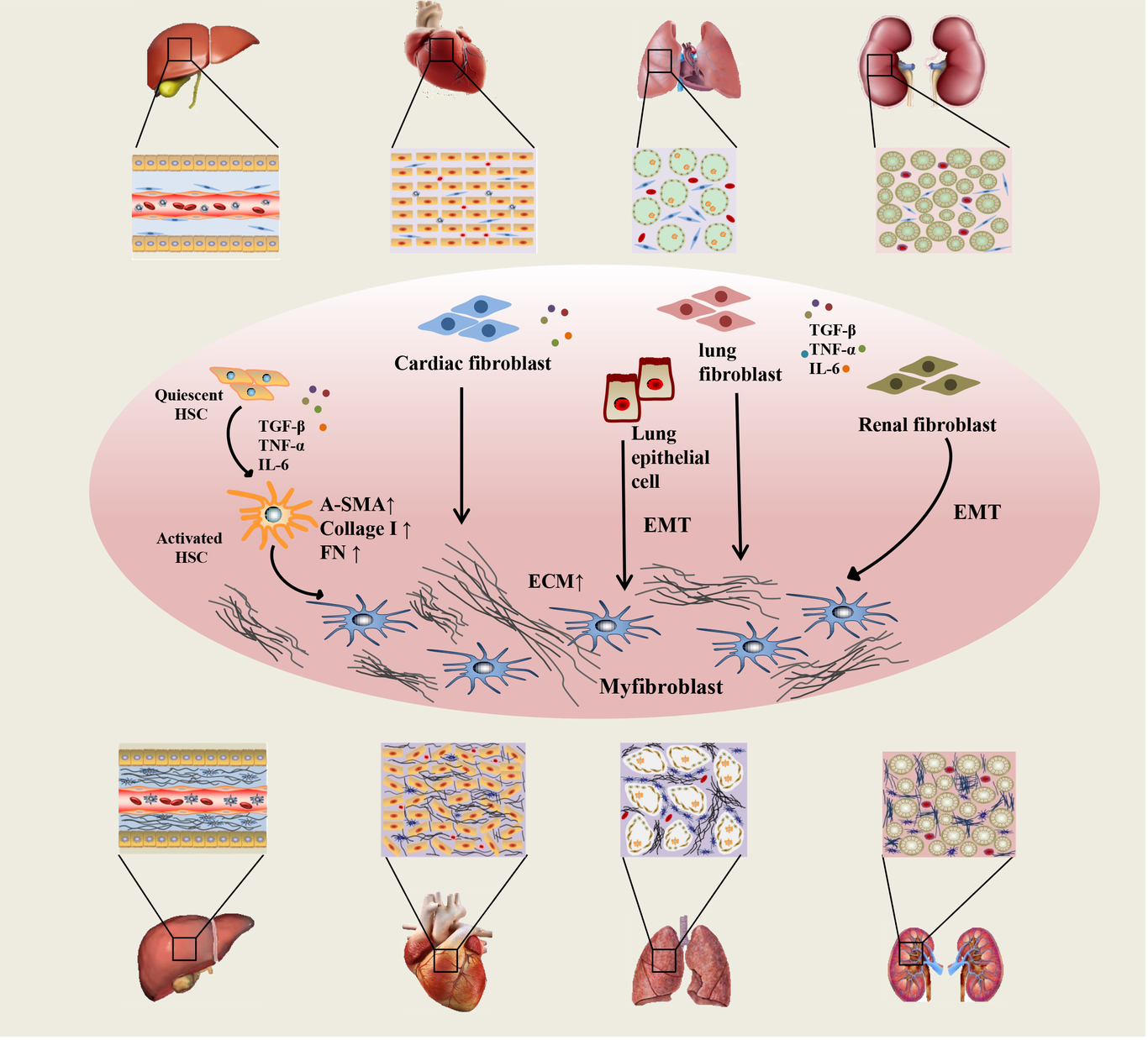
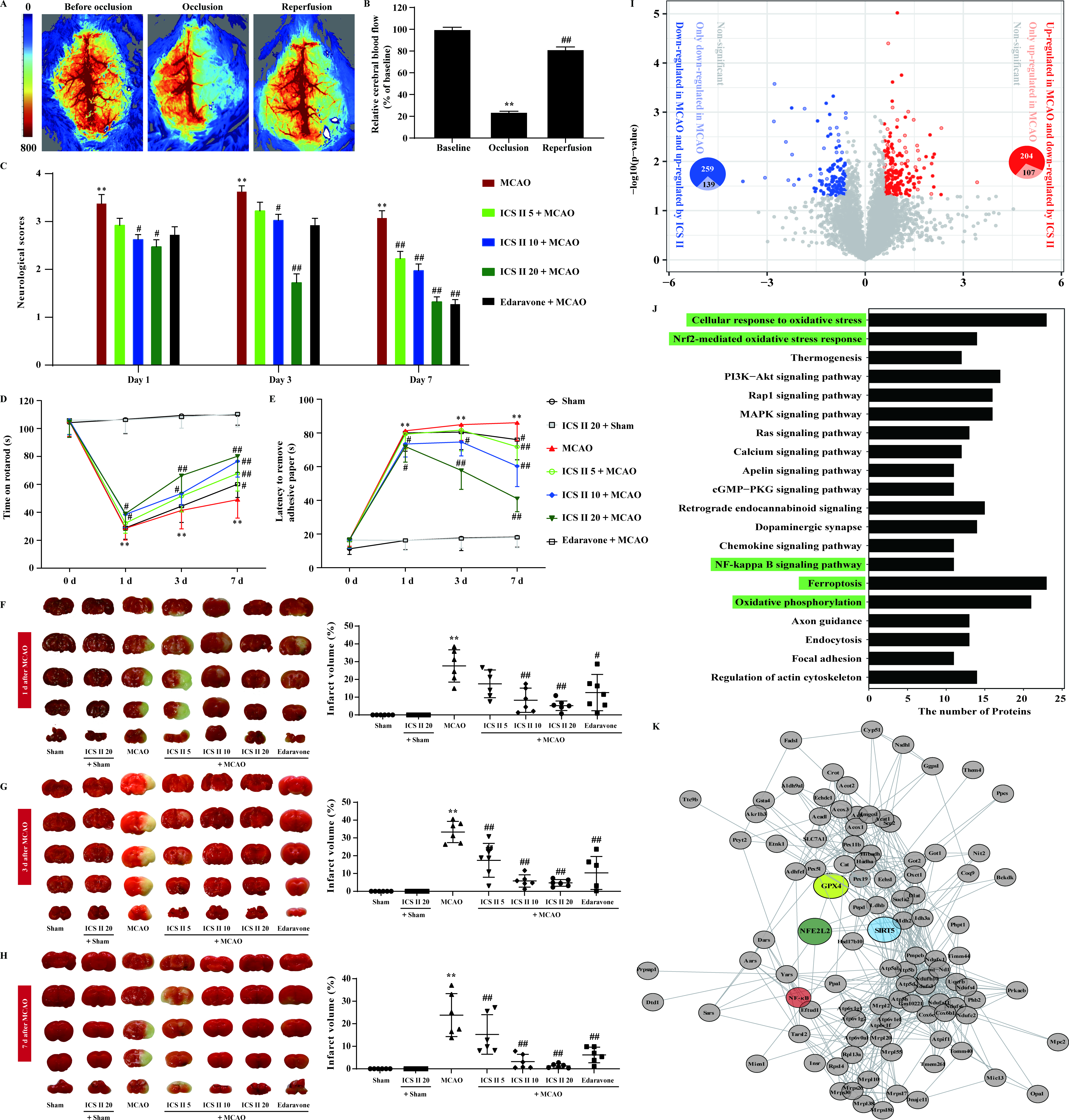
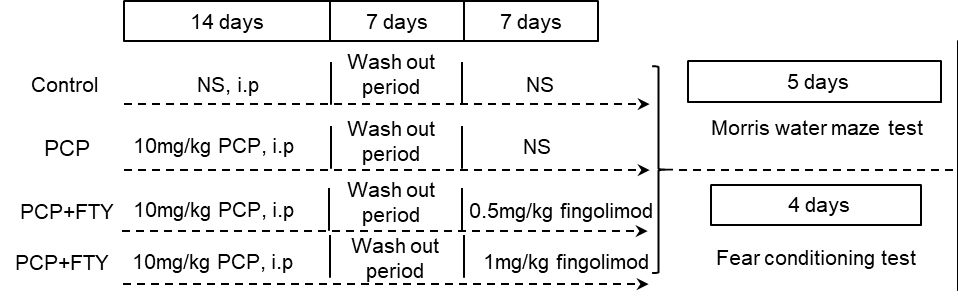
/FIbrosis mechanisms (7).png?1648618865)