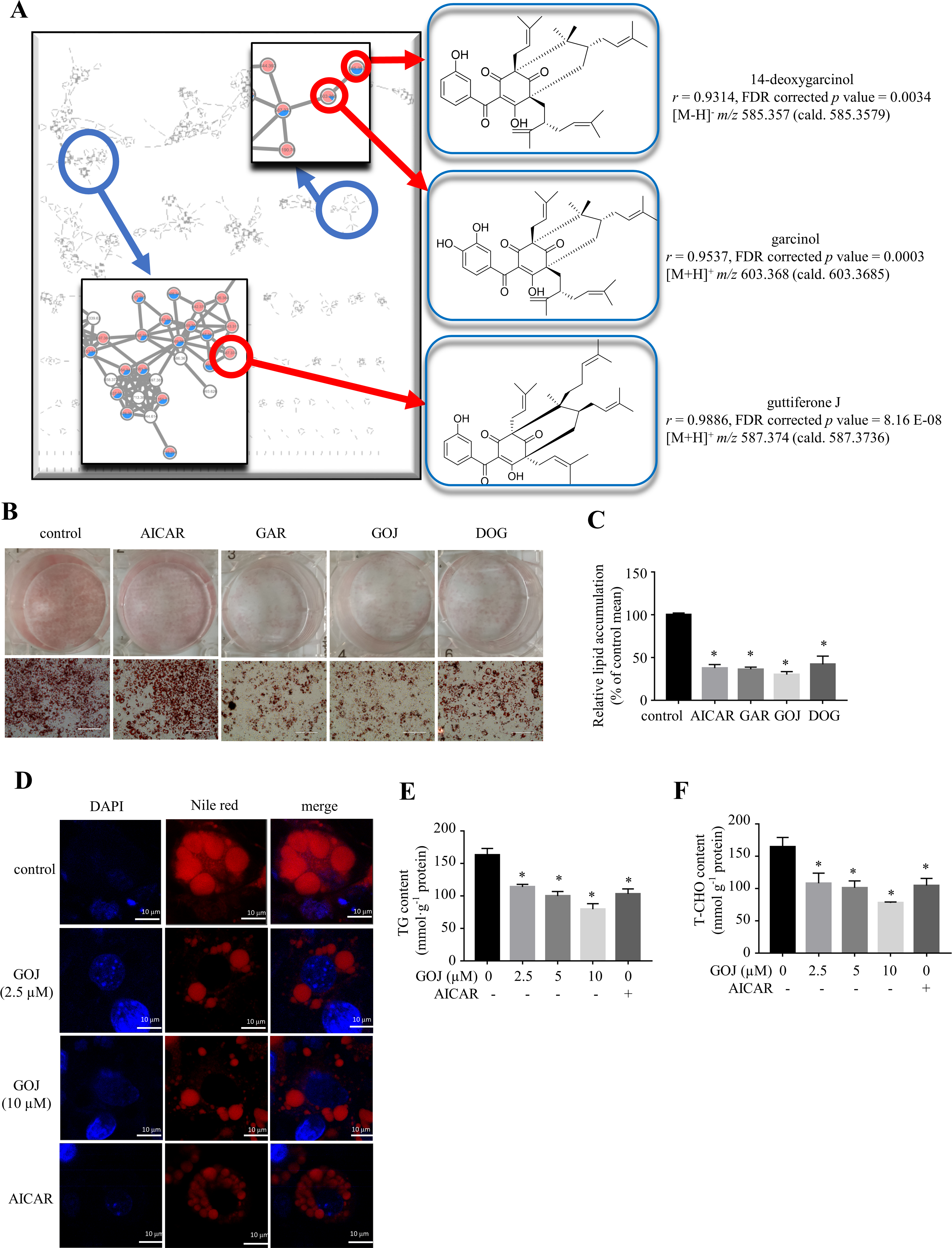
Background and Purpose: Pharmacological intervention to induce white adipose tissue browning provides a promising anti-obese therapy. The fruits of Garcinia cambogia (Clusiaceae) have been widely applied to manage body weight. The current study aims to uncover the chemical principles responsible for the anti-obese property of the fruits of G. cambogia and investigate the underlying mechanisms. Experimental Approach: The bioactivity-based molecular networking and Oil-red O staining on 3T3-L1 and C3H10T1/2 adipocytes were applied for guided isolation. High-fat diet-induced obese mice were recruited to evaluate the anti-obese activity. Key Results: Guided by the bioactivity-based molecular networking, several polycyclic polyprenylated acylphloroglucinols were targetedly isolated from the fruits of G. cambogia with lipid lowering effect on adipocytes, including guttiferone J (GOJ), garcinol and 14-deoxygarcinol. As the most potent one, GOJ (10 µM) reduced lipid accumulation by 70% and 76% in 3T3-L1 and C3H10T1/2 adipocytes, respectively. Furthermore, GOJ (2.5‒10 µM) activated the deacetylase Sirtuin 3 (SIRT3), which, in turn, reduced the acetylation level of PPARγ coactivator-1α to boost mitochondrial biogenesis, and promoted uncoupling protein 1 expression and function to enhance thermogenesis, resulting in browning of adipocytes. In high-fat diet-induced-obese mice, GOJ (10 and 20 mg∙Kg-1) protected against adiposity, hyperlipidemia, insulin resistance and liver lipotoxicity, through boosting SIRT3-mediated browning of inguinal white adipose tissue. Conclusions and Implications: The bioactivity-based molecular networking is a promising strategy for guided isolation of bioactive molecules, and GOJ represents a new scaffold of thermogenic inducer, which might be responsible for the anti-obese property of G. cambogia.


/FIbrosis mechanisms (7).png?1648618865)