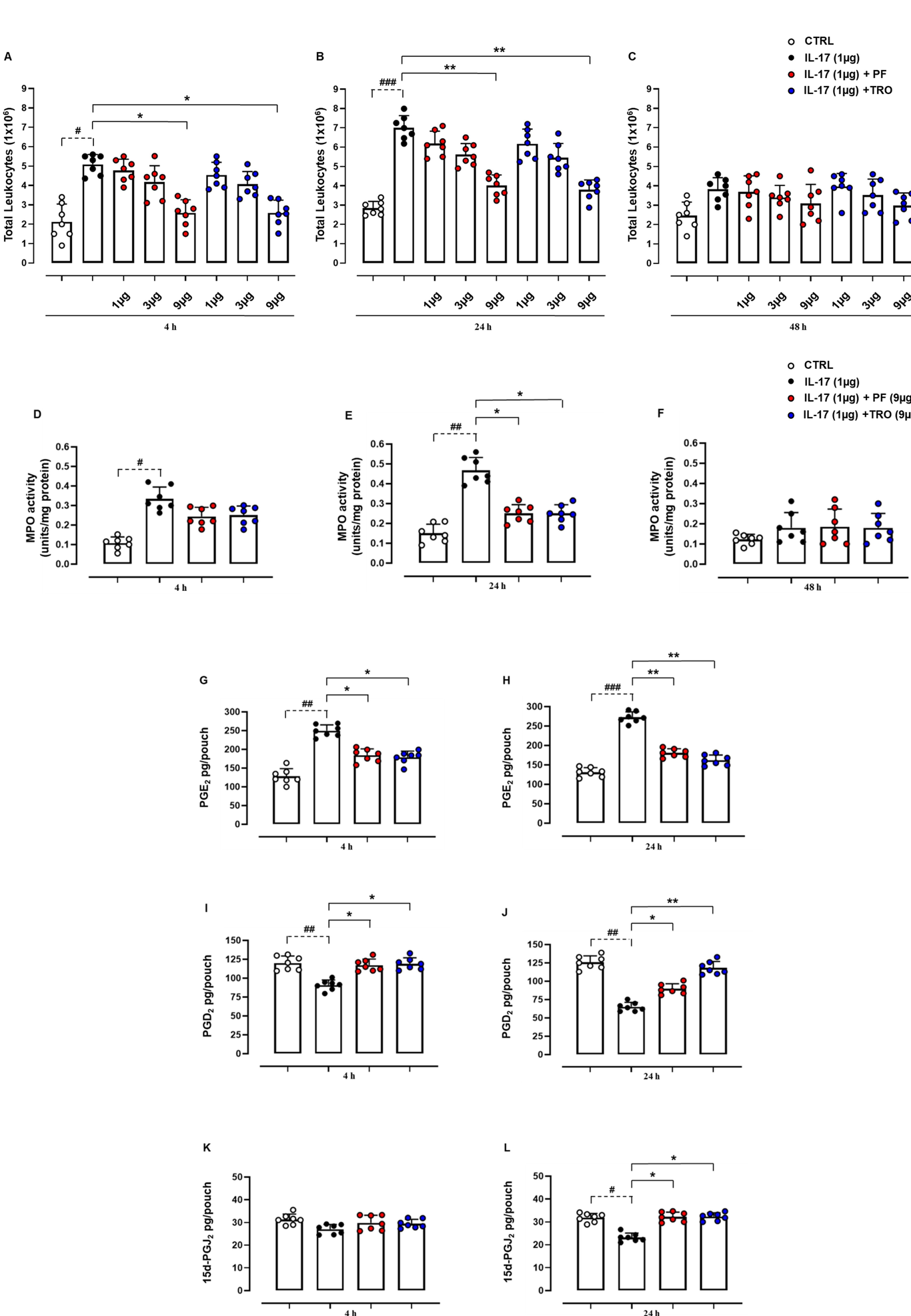
Background and Purpose: Recent biochemical and pharmacological studies have reported that in several tissues and cell types, microsomal prostaglandin E2 synthase (mPGES) and peroxisome proliferator-activated receptor-γ (PPAR-γ) expression are modulated by a variety of inflammatory factors and stimuli Considering that very little is known about the biological effects promoted by IL-17 in the context of mPGES-1/PPAR-γ modulation, we sought to investigate the contribution of this unique cytokine on these integrated pathways during the onset of inflammation. Experimental Approach: We evaluated PF 9184 (mPGES-1 antagonist) and Troglitazone (PPAR-γ agonist) activity utilising integrated in vitro and in vivo approaches. The dorsal air pouch model was employed, and inflammatory infiltrates were analysed by flow cytometry. Locally produced cyto-chemokines and prostaglandins were assessed using ELISA assays. Western blots were also employed to determine the activity of various enzymes involved in downstream signalling pathways. Key Results: PF 9184 and Troglitazone, in a time and dose-dependent manner, were shown to significantly modulate leukocyte infiltration, myeloperoxidase activity, and the expression of COX-2/mPGES-1, NF-кB/IкB-α and mPGDS-1/PPAR-γ induced by IL-17. Moreover, both compounds were found to modulate prostaglandins (PGE2, PGD2, and PGJ2) production, the expression of different pro-inflammatory cyto-chemokines and the recruitment of inflammatory monocytes in response to IL-17. Conclusions and Implications: Collectively, the data presented suggests that IL-17 may constitute a specific modulator of inflammatory monocytes during later phases of the inflammatory response. Therefore, the results of this study show, for the first time, that IL-17/mPGES-1/PPAR-γ “axis” could represent a potential therapeutic target for inflammatory-based and immune-mediated diseases.
/Figure 1 (Saier and Peyruchaud).png?1611463666)