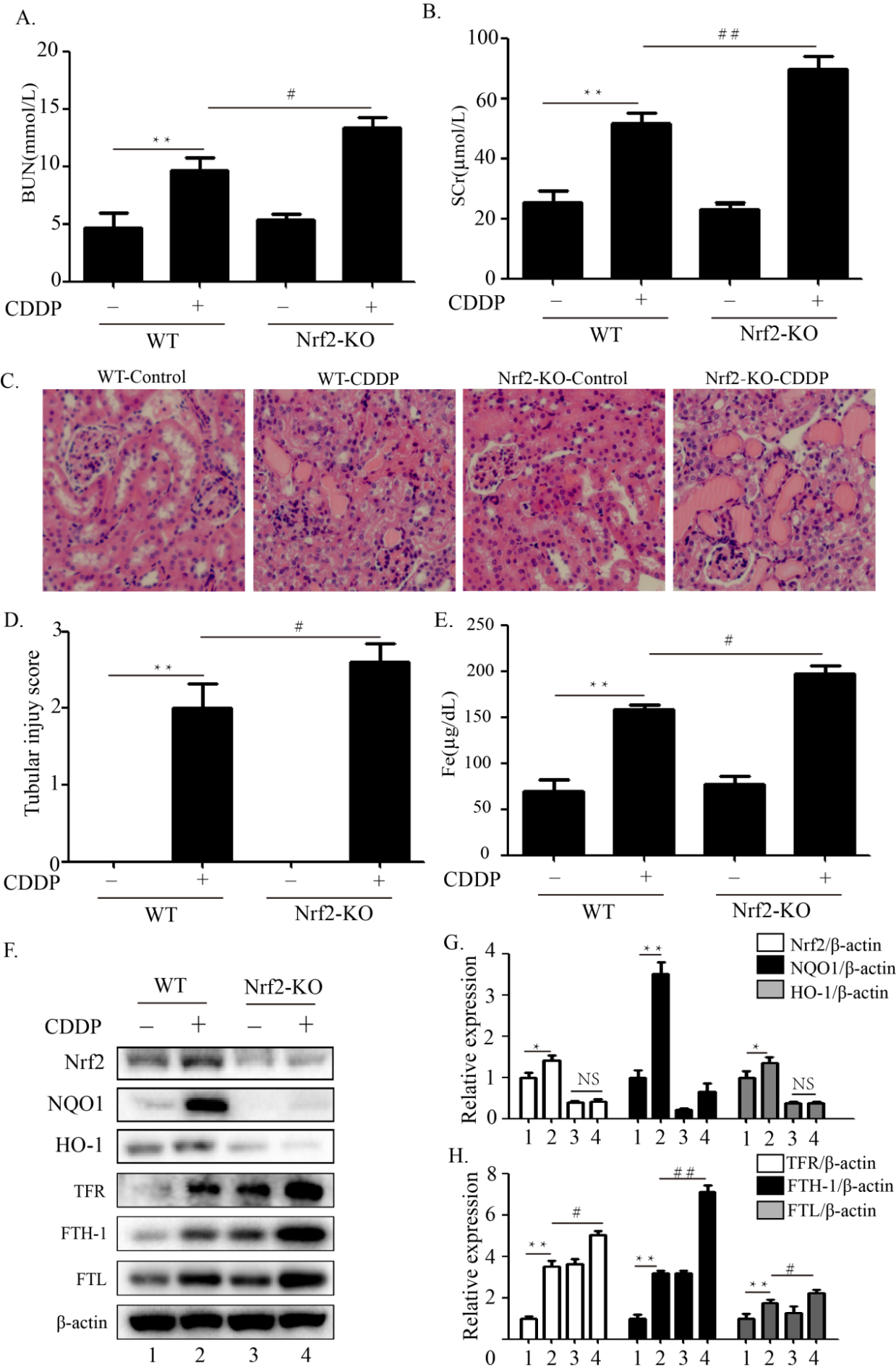
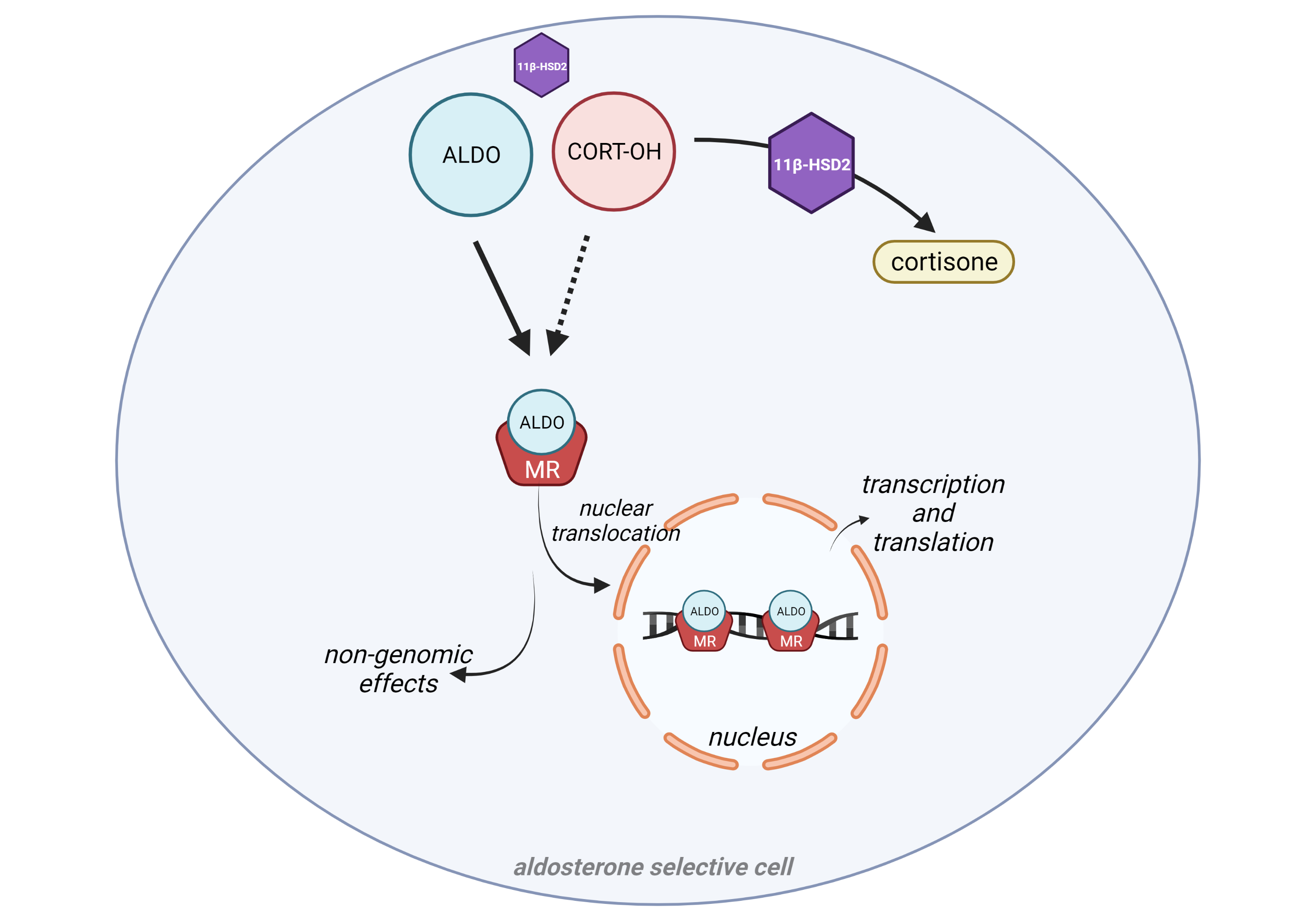
In the retina, mineralocorticoid receptor (MR), expressed in vessels, glial and neuronal cells, is mainly activated by glucocorticoids. Under pathological conditions, ocular MR expression and corticoids change, leading in most cases to MR overactivation. Experimental models using MR agonists or antagonists, administered systemically or intraocularly, acutely or chronically and transgenic models, allowed to identify the deleterious consequences of MR pathway overactivation. Among them, oxidative stress, inflammation, deregulation of hydro-ionic channels, alteration of choroidal vasculature, angiogenesis and cell death, are common to major retinal diseases. Specific MR antagonists showed efficacy in models of diabetic retinopathy, ischaemia, retinal and choroidal angiogenesis and in models of glaucoma. It is highly likely that MR antagonists will find a place in the therapeutic arsenal of age-related macular degeneration, diabetic retinopathy, glaucoma and in pachychoroid associated diseases. Their use in humans is still limited by the need of biomarkers of MR activation and specific ocular formulations.