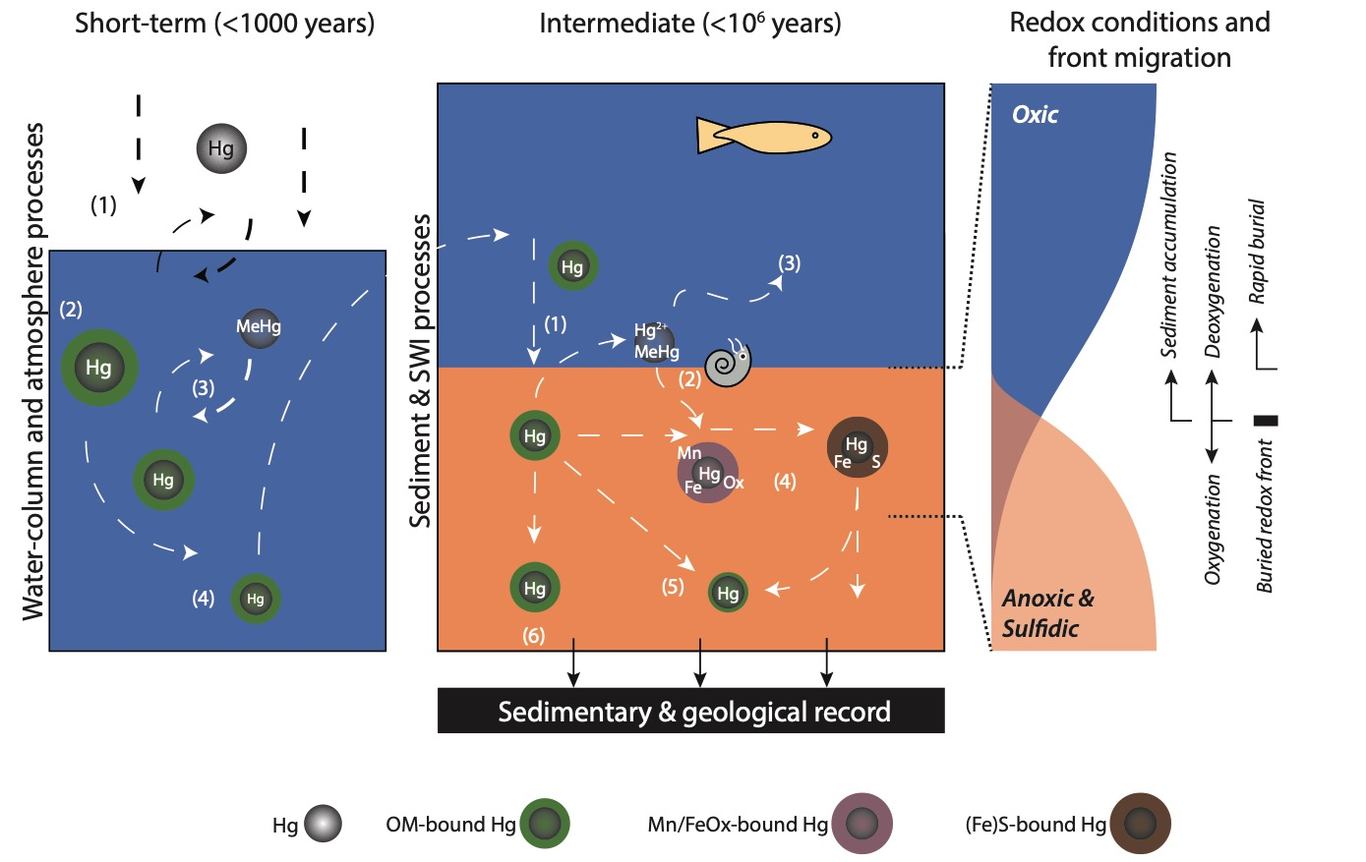
Volcanism is the dominant natural source of mercury (Hg) to the atmosphere, biosphere, ocean and sediments. In recent years, sedimentary Hg contents have emerged as a tool to reconstruct volcanic activity, and particularly activity of (subaerially emplaced) large igneous provinces (LIP) in geological deep time. More specifically, Hg has shown potential as a useful proxy to illuminate the previously elusive impact of such large-scale volcanism on marine and terrestrial paleo-environments. While Hg is now widely applied as volcanism tracer, non-volcanic factors controlling sedimentary Hg content are generally not well constrained. Part of this uncertainty stems from our inability to directly observe a natural unperturbed “steady-state” environment as a baseline, as the modern Hg cycle is heavily influenced by anthropogenic activity. Here we focus on the effects of ambient redox conditions in the water column and shallow sediments (early diagenesis), quantify their influence on the geological Hg record and thereby constrain their potential impact on the use of Hg as a proxy for deep-time volcanic activity. Constraining these factors is of critical importance for the application of Hg as a proxy. Many periods in the geological past for which records have been generated, such as the Mesozoic Oceanic Anoxic Events, are marked by a variety of high-amplitude environmental perturbations, including widespread deoxygenation and deposition of organic-rich sediments. We estimate the impact of redox changes and early diagenesis on the geological Hg record using a suite of (sub)recent–Pleistocene and Upper Cretaceous sediments representing oxic to euxinic marine conditions. Our sample set includes a transect through an oxygen minimum zone and cores that record transient shifts in oxygenation state, as well as post-depositional effects – all unrelated to volcanism, to the best of our knowledge. We find substantial alterations to the Hg record and the records of organic carbon and total sulfur, which are typically assumed to be the most common carrier phases of Hg in marine sediments. Moreover, these biases can lead to signal-alterations on a par with those interpreted to result from volcanic activity. Geochemical modifications are ubiquitous and their potential magnitude implies that the factors leading to biases in the geological record warrant careful consideration before interpretation. Factors of particular concern to proxy application are (1) the disproportionate loss of organic carbon and sulfur compounds relative to Hg during oxidation that strongly modulates normalized Hg records, (2) the evasion of Hg in anoxic and mildly euxinic sediments and (3) sharp focusing of Hg during post-depositional oxidation of organic matter.