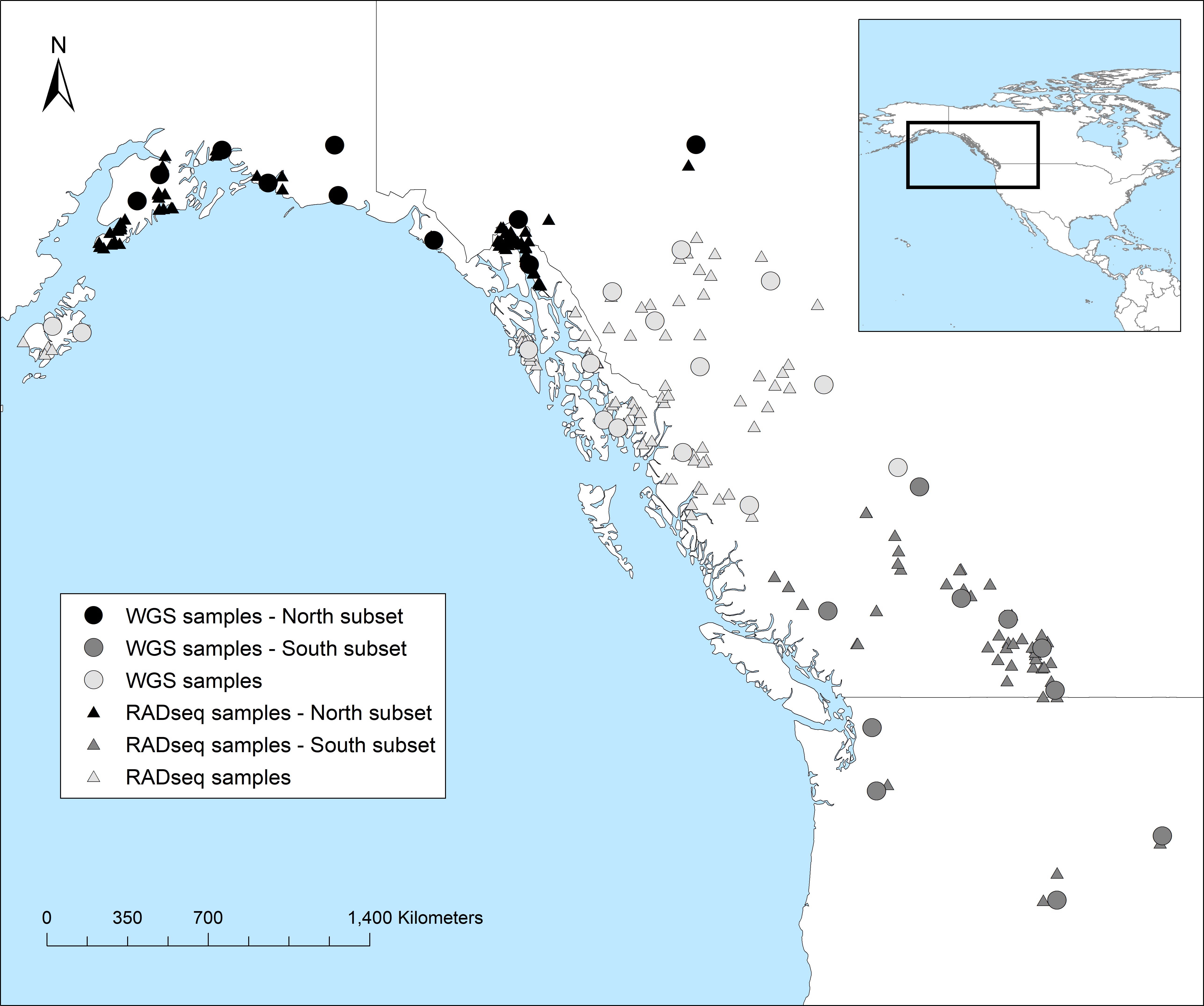
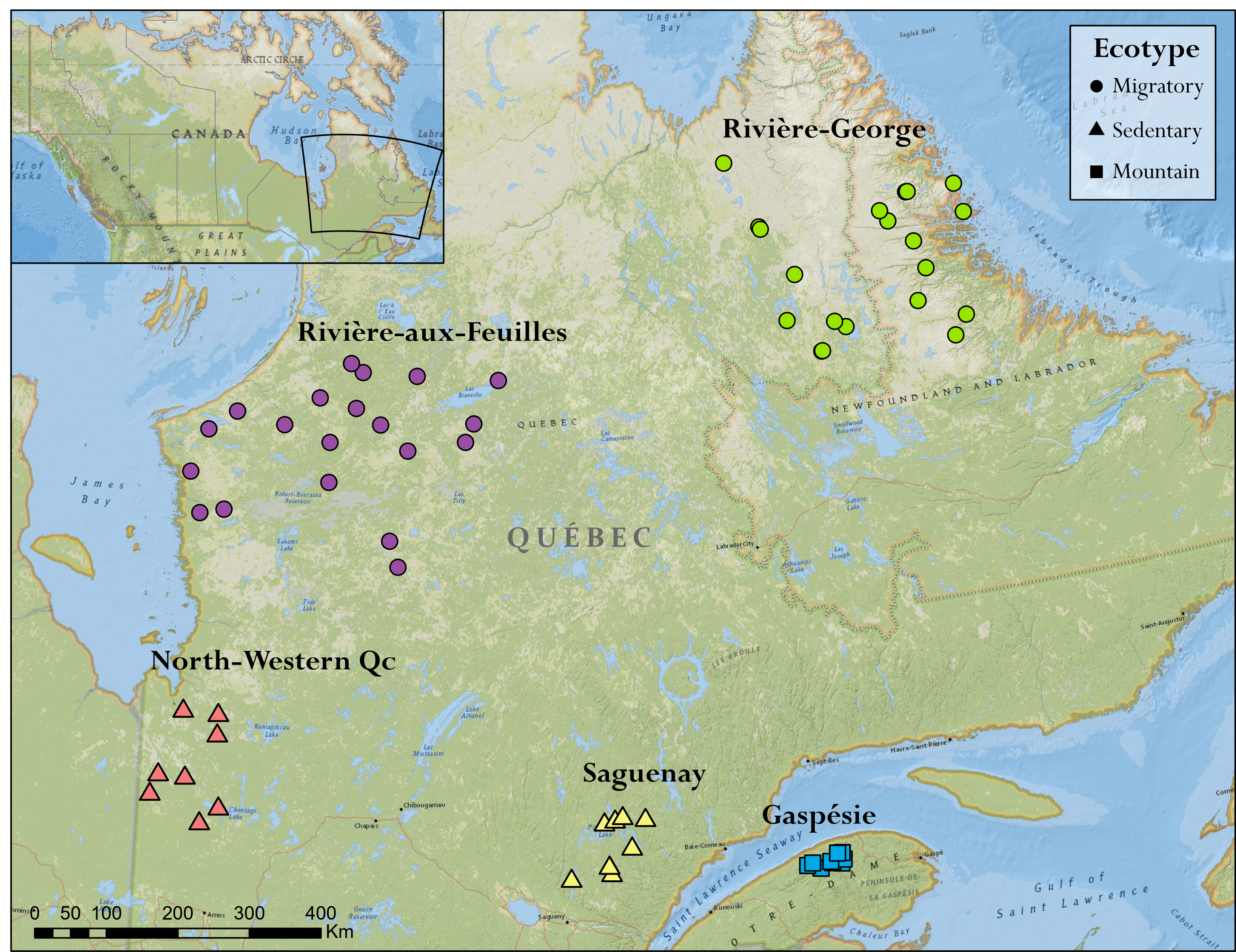
The loss of genetic diversity is a challenge many species are facing, and genomics is a potential tool that can inform and prioritize decision making. Caribou populations have experienced significant recent declines throughout Québec, Canada, and some are considered threatened or endangered. We calculated the ancestral and contemporary patterns of genomic diversity of five caribou populations and applied a comparative framework to assess the interplay between demography and genomic diversity. We calculated a caribou specific mutation rate, μ, by extracting orthologous genes from related ungulates. Whole genome re-sequencing was completed on 67 caribou and genotype likelihoods were estimated. We calculated nucleotide diversity, θπ and estimated the coalescent or ancestral Ne, which ranged from 12,030 to 15,513. When compared to the census size, NC, the endangered Gaspésie Mountain caribou population had the highest Ne:NC ratio which is consistent with recent work suggesting high ancestral Ne:NC is of conservation concern. These ratios were highly correlated with genomic signatures (i.e. Tajima’s D) and explicit demographic model parameters. Values of contemporary Ne, estimated from linkage-disequilibrium, ranged from 11 to 162, with Gaspésie having among the highest contemporary Ne:NC ratio. Importantly, conservation genetics theory would predict this population to be of less concern based on this ratio. Of note, F varied only slightly between populations, and runs of homozygosity were not abundant in the genome. Our study highlights how genomic patterns are nuanced and misleading if viewed only through a contemporary lens; a holistic view should integrate ancestral Ne and Tajima’s D into conservation decisions.