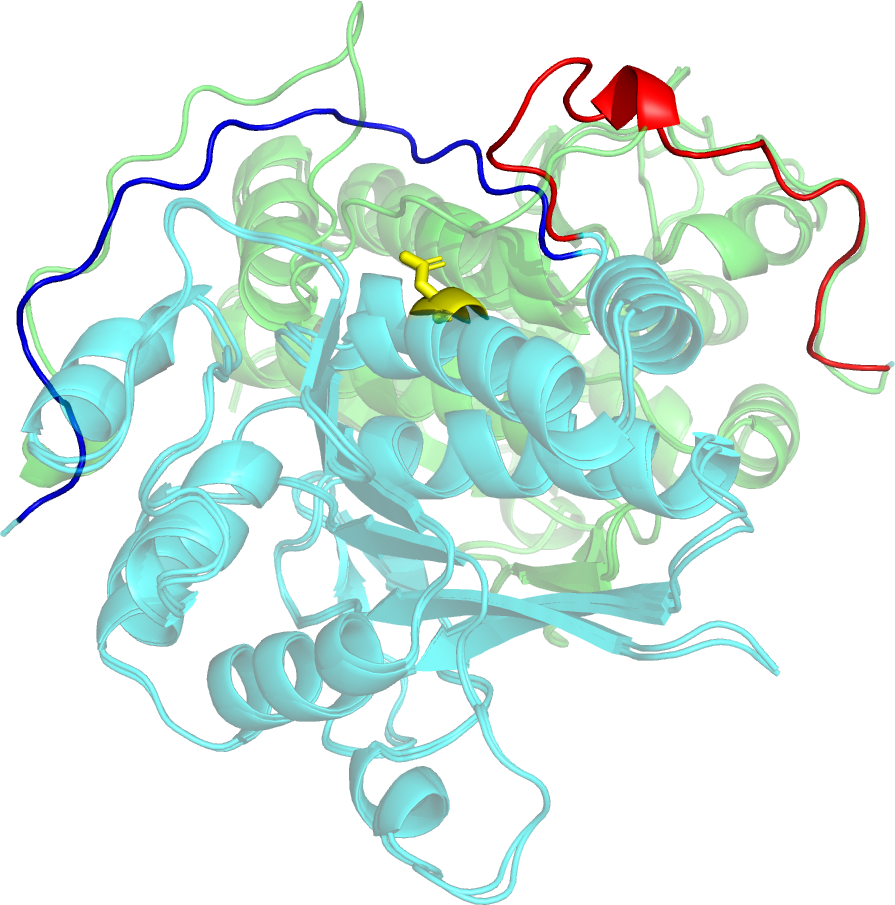
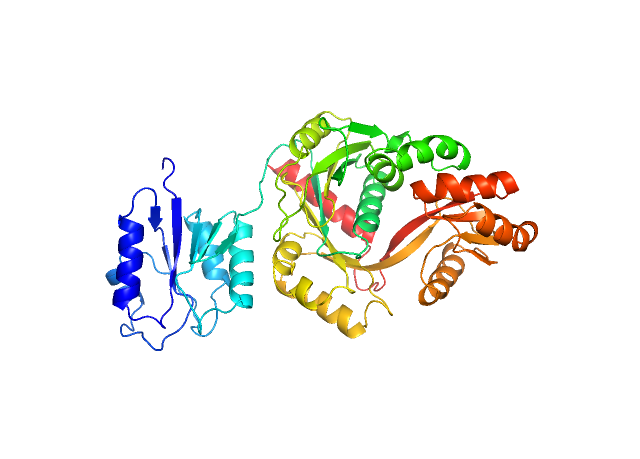
The results of tertiary structure assessment at CASP15 are reported. For the first time, recognising the outstanding performance of AlphaFold 2 (AF2) at CASP14, all single chain predictions were assessed together, irrespective of whether a template was available. At CASP15 there was no single stand-out group, with most of the best-scoring groups - led by PEZYFoldings, UM-TBM and Yang Server - employing AF2 in one way or another. Many top groups paid special attention to generating deep Multiple Sequence Alignments (MSAs) and testing variant MSAs, thereby allowing them to successfully address some of the hardest targets. Such difficult targets, as well as lacking templates, were typically proteins with few homologues: small size, high α-helical content and monomeric structure were other likely aggravating factors. Local divergence between prediction and target correlated with localisation at crystal lattice or chain interfaces, and with regions exhibiting high B-factor factors in crystal structure targets, but should not necessarily be considered as representing error in the prediction. However, analysis of exposed and buried side chain accuracy showed room for improvement even in the latter. Nevertheless, a majority of groups, including those applying methods similar to those used to generate major resources such as the AlphaFold Protein Structure Database and the ESM Metagenomic atlas, produced high quality predictions for most targets which are valuable for experimental structure determination, functional analysis and many other tasks across biology.