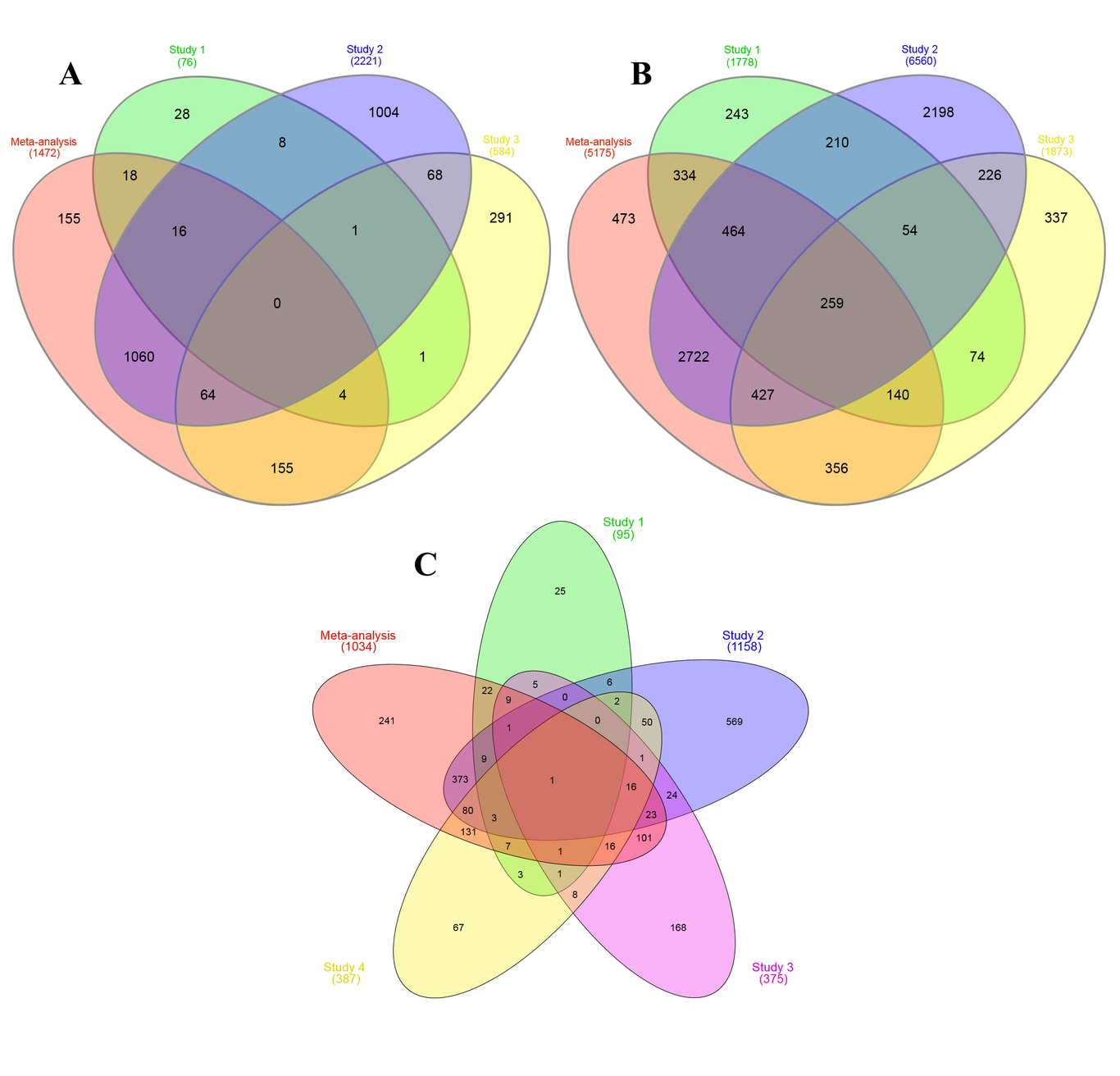
Published article in the link below:https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-023-09673-yAbstractBackground: Olive oil contains monounsaturated oleic acid up to 83% and phenolic compounds, making it an excellent source of fat. Due to its economic importance, the quantity and quality of olive oil should be improved in parallel with the international standards. In this study, we analyzed the raw RNA-seq data with a meta-analysis approach to identify important genes and their metabolic pathways involved in olive oil quality. Results: A deep search of RNA-seq published data shed light on thirty experiments associated with the olive transcriptome, four of these proved to be ideal for meta-analysis. Meta-analysis confirmed the genes identified in previous studies and released new genes, which were not identified before. According to the IDR index, the meta-analysis had good power to identify new differentially expressed genes. The key genes were investigated in the metabolic pathways and were grouped into four classes based on the biosynthetic cycle of fatty acids and factors that affect oil quality. Galactose metabolism, glycolysis pathway, pyruvate metabolism, fatty acid biosynthesis, glycerolipid metabolism, and terpenoid backbone biosynthesis were the main pathways in olive oil quality. In galactose metabolism, raffinose is a suitable source of carbon along with other available sources for carbon in fruit development. The results showed that the biosynthesis of acetyl-CoA in glycolysis and pyruvate metabolism is a stable pathway to begin the biosynthesis of fatty acids. Key genes in oleic acid production as an indicator of oil quality and critical genes that played an important role in production of triacylglycerols were identified in different developmental stages. In the minor compound, the terpenoid backbone biosynthesis was investigated and important enzymes were identified as an interconnected network that produces important precursors for the synthesis of a monoterpene, diterpene, triterpene, tetraterpene, and sesquiterpene biosynthesis.Conclusions: The results of the current investigation can produce functional data related to the quality of olive oil and would be a useful step in reducing the time of cultivar screening by developing gene specific markers in olive breeding programs, releasing also new genes that could be applied in the genome editing approach.IntroductionOlive oil is a particularly important product because of its fatty acids and phenolic compounds, which are mainly responsible for the beneficial health aspects. Among these compound, high oleic acid content and the presence of minor bioactive compounds are the reason for its attribution as the healthiest among all vegetable oils (Guclu et al., 2020; Lozano-Castellón et al., 2020; Yubero-Serrano et al., 2019). The International Olive Council (IOC) has also stated that oils with the highest levels of oleic acid are the most valuable nutritional products (IOC, 2015). Currently, the cultivation areas and oil production increased in the world but only a few cultivars can yield consistently in the new environmental conditions and often changing negatively their quality profiles (Borges et al., 2017; García-Inza et al., 2016; Torres et al., 2017). Therefore, according to the economic importance of olive oil, the quantity and quality of oil should be improved in parallel, based on international standards. Several studies have noted that the main factors that influence olive oil quality are genotype, climatic and agronomic conditions, edaphic factors, and the technological method applied for oil extraction. Among these factors, genotype has a preponderant influence (Ambra et al., 2017; Baiano et al., 2013; Beltrán et al., 2016; de la Rosa et al., 2016; Mele et al., 2018; Miho et al., 2021; Mikrou et al., 2020; Pérez et al., 2018; Rugini et al., 2016). Besides studies show that 70% of the observed diversity in terms of fatty acid composition, phenolic compounds, bitterness or taste, and oil stability is genetically influenced (Mousavi et al., 2019, 2022; Parvini et al., 2015; Riachy et al., 2019).One of the most important goals of RNA-seq experiments is to investigate changes in gene expression profiles under two or more different experimental conditions. The most of the RNA-seq studies performed in olives were related to the study of biotic and abiotic stresses (Grasso et al., 2017; Nissim, Shlosberg, et al., 2020), micro-RNA identification (Yanik et al., 2013), fruit developmental stages (Alagna et al., 2009, 2016; Galla et al., 2009; Guodong et al., 2019), and cold acclimation (De La O Leyva-Pérez et al., 2015; Guerra et al., 2015). Recently, some studies have focused on the role of environmental stresses such as high temperature and the altitude of cultivated areas on oil content and its quality using RNA-seq technique, but a limited number of these studies were directly related to the evaluation of oil quality (Bruno et al., 2019; Nissim, Shloberg, et al., 2020; Nissim, Shlosberg, et al., 2020). In a study performed by Galla et al. (2009), suppression subtractive hybridization (SSH) was used to isolate and identify a large set of genes that were differentially expressed at three different stages along olive fruit development. In another study conducted by Alagna et al., 2009, differentially expressed genes involved in the metabolism of phenol and fatty acids at different stages of olive fruit development were identified. Moreover, in 2013, the olive cultivars' transcripts were used for de novo assembly and functional annotation (Rodríguez, 2013). In a study conducted by Parra et al. (2013), the transcriptional regulation of the ripening process and activation of abscission zone were detected by RNA-seq. In 2016, the genome of Farga was sequenced and its annotation was identified by RNA-seq of leaf, root, and fruit samples (Cruz et al., 2016). Considering the importance of phenols in olive, in 2016, de novo transcriptome assembly was reported in olive fruit at different development stages and transcripts involved in flavonoid and anthocyanin pathways were identified (Iaria et al., 2016). In another study (Unver et al., 2017), the wild olive genome was sequenced and transcriptome analysis was performed to identify genes involved in oil biosynthesis. Recently, transcripts of all the enzymes in the biosynthetic pathway of tyrosol, hydroxytyrosol and secologanin, oleuropein's precursor, were identified by the RNA-seq method in Koroneiki cultivar (Mougiou et al., 2018). Furthermore, in 2019, targeted metabolome, Pacbio ISOseq transcriptome, and Illumina RNA-seq transcriptome were combined to investigate the relationship between phenols biosynthesis and differentially expressed genes during olive fruit development (Guodong et al., 2019). Technical variation in different experiments could affect the reproducibility of the research. Moreover, due to the cost of sequencing, RNA-seq experiments are mainly performed in a limited number of biological replicates, reducing the statistical power and the ability to detect and validate differences in gene expression. Accordingly, one of the most effective ways to improve reproducibility is to use multiple datasets through meta-analysis (Keel et al., 2018). Therefore, re-analyzing existing data derived from several independent experiments can reveal new information and evaluate the most reliable key genes in a certain biosynthetic pathway. Meta-analysis of RNA-seq data can increase the speed of production of functional data related to the quality of olive oil and produce useful information. This study was conducted to analyze RNA-seq data obtained from multiple studies by meta-analysis approach to validate and identify key genes involved in the main metabolic pathways of oil quality. ResultsThe SRA and literature searches results showed that there were thirty experiments associated with the olive transcriptome. By applying the filtration and quality control reported in the methods section, four experiments had the ideal conditions to enter in our meta-analysis. The meta-analysis was performed to compare growth stages in pairs (C1, C2, and C3) and the results of each comparison were shown as an independent Venn diagram (Figure 1). The meta-analysis individuates 1472 differential expressed genes in C1 comparison from which, 155 differential expressed genes were identified for the first time in the present study (Figure 1A). The C2 comparison has identified 5175 differential expressed genes (Figure 1B), among them, 473 differential expressed genes have never been reported in the previous studies. The PRJNA260808 had only two developmental stages, S2 and S3, so only the C3 comparison was considered in this experiment. The results of C3 comparison identified 1034 differential expressed genes (Figure 1C), and in addition, 241 of them were identified for the first time.The ratio of the identified differentially expressed genes in the meta-analysis to the total identified differential expressed genes were calculated as Integration-driven Discovery Rate (IDR) for each comparison. The IDR for C1 was 10.53 and in C2 and C3 were 9.14 and 23.13, respectively.