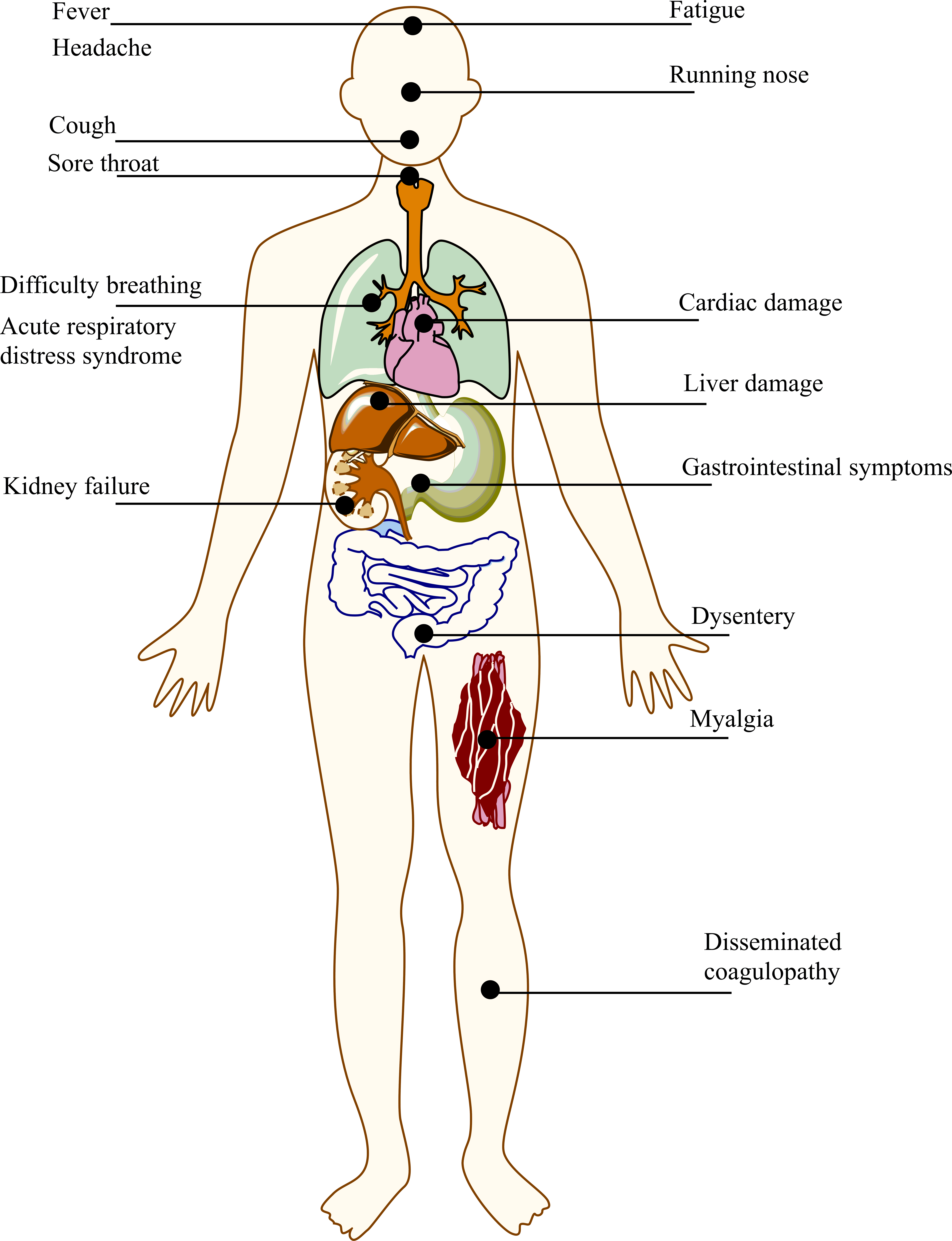
IImmunodeficiency and hyperinflammation characterize COVID-19 associated states; thus, repurposing of multiple cytokine and/or anti-cytokine drugs currently being used in other therapeutic areas has been suggested as a potential therapeutic strategy in COVID-19 patients. Clinical trials involving these drugs target the most frequent and life-threatening peripheral consequences of the disease, mainly focusing on lung, heart, and coagulation functions; however, a growing number of reports describe a wide range of COVID-associated neurological manifestations (altogether defined as neuro-COVID) including anosmia, seizures, confusion, stroke, encephalopathy, and paralysis. Notably, the underlying pathophysiological mechanisms for neuro-COVID may also include dysregulation of cytokines/chemokines, deficiencies in the innate immune response, and autoimmunity. This suggests that therapeutic attempts with drugs targeting cytokine-mediated inflammation in peripheral organs could also positively affect neuro-COVID manifestations. As a matter of fact, some of these drugs have also been scrutinized for their potential efficacy in treating neuroinflammatory diseases such as optic neuromyelitis, epilepsy, stroke, and traumatic brain injury, among others. On the other hand, anti-cytokine drugs, by impairing relevant physiological activities exerted by these mediators in the CNS, may also be endowed with significant neurological risk. Therefore, the primary aim of the present manuscript is to review the available preclinical and clinical data regarding the neurological effects of the drugs targeting cytokine-mediated inflammation, in order to raise awareness about their potentially beneficial or detrimental neurological consequences when used to treat COVID-19 patients.