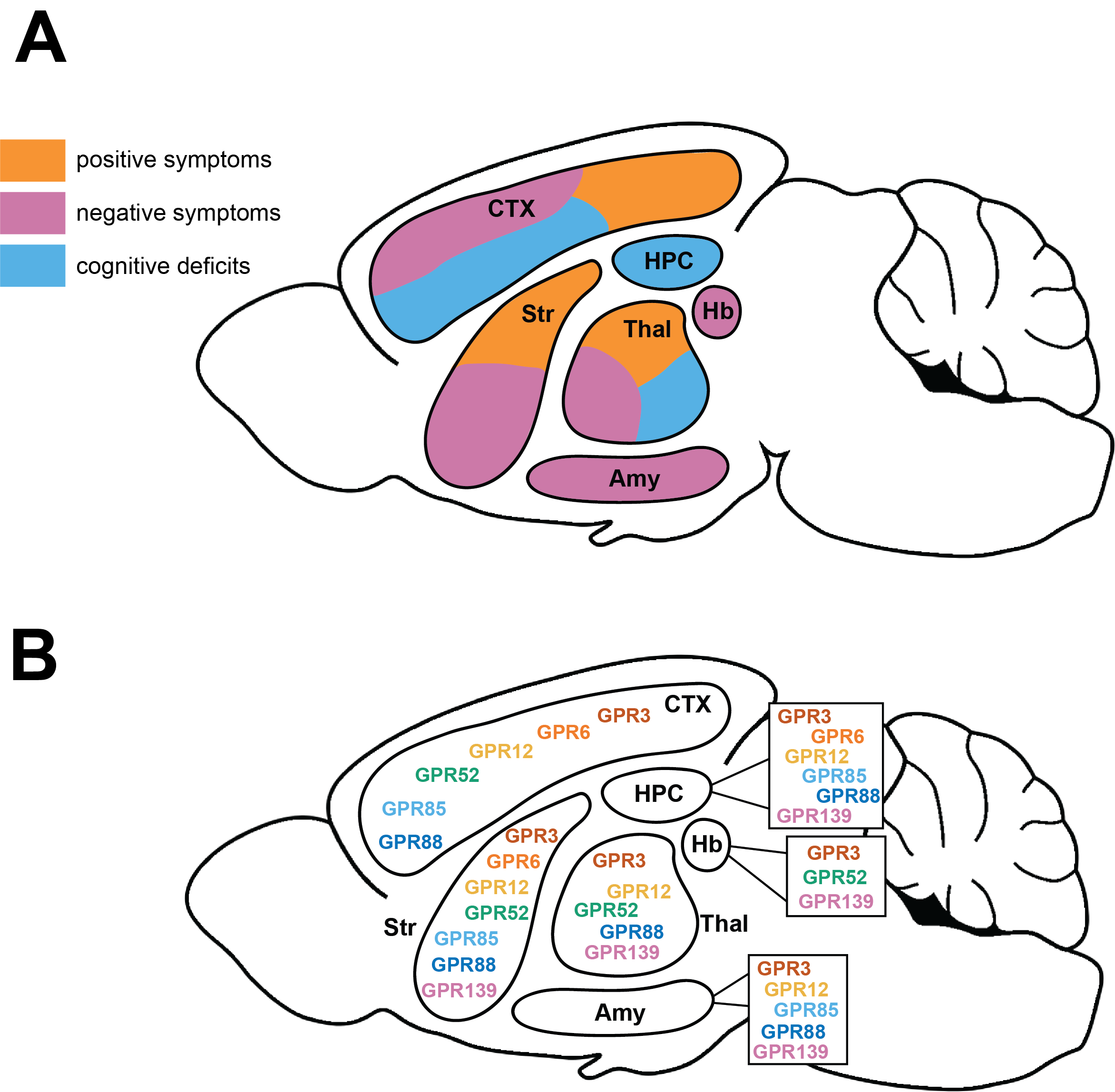
Schizophrenia remains a sizable socioeconomic burden that continues to be treated with therapeutics based on 70-year old science. All currently approved therapeutics primarily target the dopamine D2 receptor to achieve their efficacy. Whilst dopaminergic dysregulation is a key feature in this disorder, the targeting of dopaminergic machinery has yielded limited efficacy and an appreciable side effect burden. Over the recent decades, numerous drugs that engage non-dopaminergic GPCRs have yielded a promise of efficacy without the deleterious side effect profile, yet none have successfully completed clinical studies and progressed to the market. More recently, there has been increased attention around non-dopaminergic GPCR-targeting drugs, notably KarXT, which demonstrated efficacy in some schizophrenia symptom domains. This provides renewed hope that effective schizophrenia may lay outside of the dopaminergic space. Despite the potential for muscarinic receptor- (and other well-characterised GPCR families) targeting drugs to treat schizophrenia, they are often plagued with complications such as lack of receptor subtype selectivity and peripheral on-target side-effects. Orphan GPCR studies have opened a new avenue of exploration with many demonstrating schizophrenia-relevant mechanisms and a favourable expression profile, thus offering potential for novel drug development. This review discusses centrally-expressed orphan G protein-coupled receptors: GPR3, GPR6, GPR12, GPR52, GPR85, GPR88 and GPR139 and their relationship to schizophrenia. We review their expression, signalling mechanisms and cellular function, in conjunction with small molecule development and structural insights. We seek to provide a snapshot of the growing evidence and development potential of new classes of schizophrenia therapeutics.