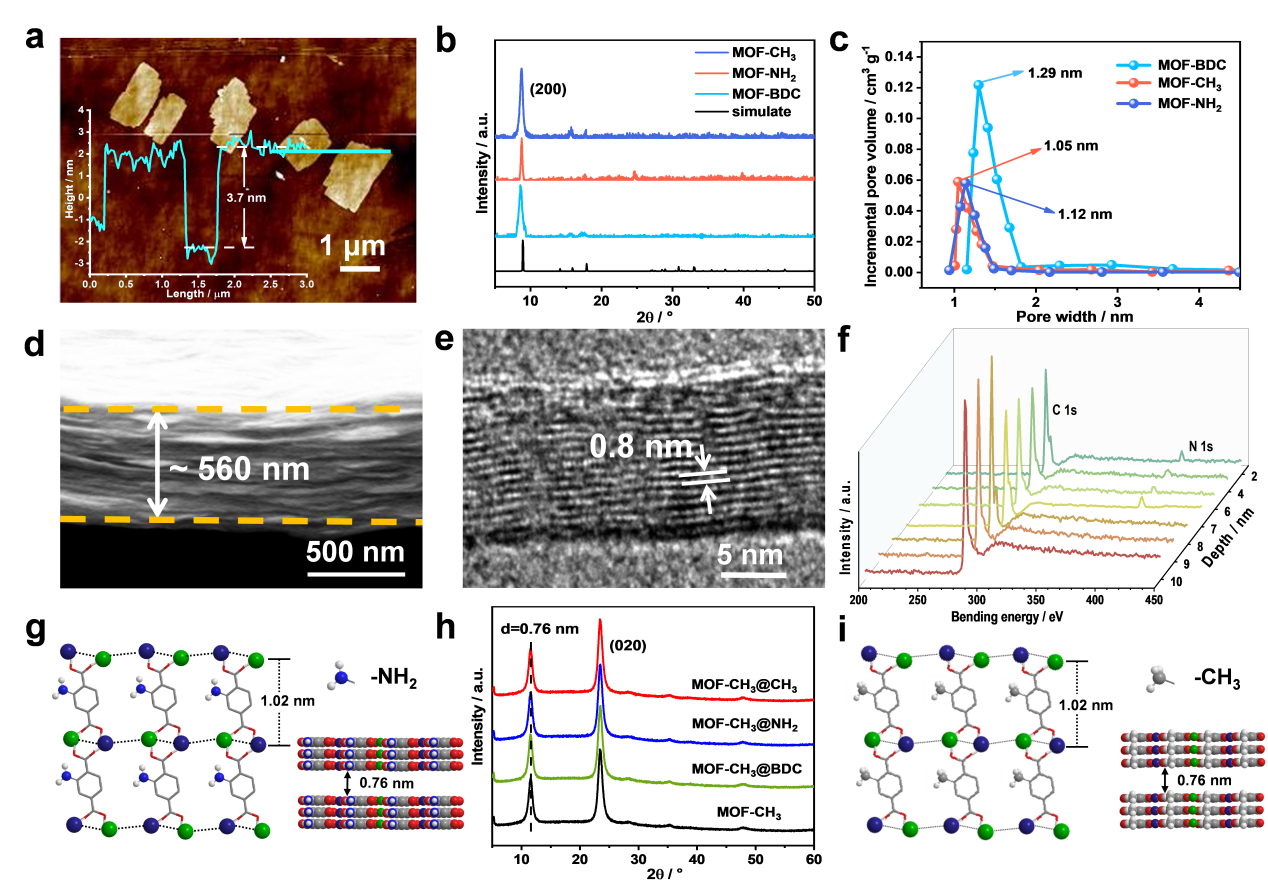
Lamellar membranes, especially assembled by microporous framework nanosheets, have excited interest for fast molecular permeation. However, the underlying molecular dissolution behaviors on membrane surface, especially at pore entrances, remain unclear. Here, hierarchical metal-organic framework (MOF) lamellar membranes with 7 nm-thick surface layer and 553 nm-thick support layer are prepared. Hydrophilic (–NH2) or hydrophobic (–CH3) groups are decorated at pore entrances on surface layer to manipulate wettability, while –CH3 groups on support layer provide comparable, low-resistance paths. We demonstrate that molecular dissolution behaviors are determined by molecule-molecule and molecule-pore interactions, derived from intrinsic parameters of molecule and membrane. Importantly, two dissolution model equations are established: for hydrophobic membrane surface, dissolution activation energy (ES) obeys ES=Kmln[(γL-γC)μd2], while turns to ES=Kaln[(γL-γC)δeμd2] for hydrophilic one. Particularly, hydrophilic pore entrances exert strong interaction with polar molecules, thus compensating the energy consumed by molecule rearrangement, giving fast permeation (> 270 L m-2 h-1 bar-1).