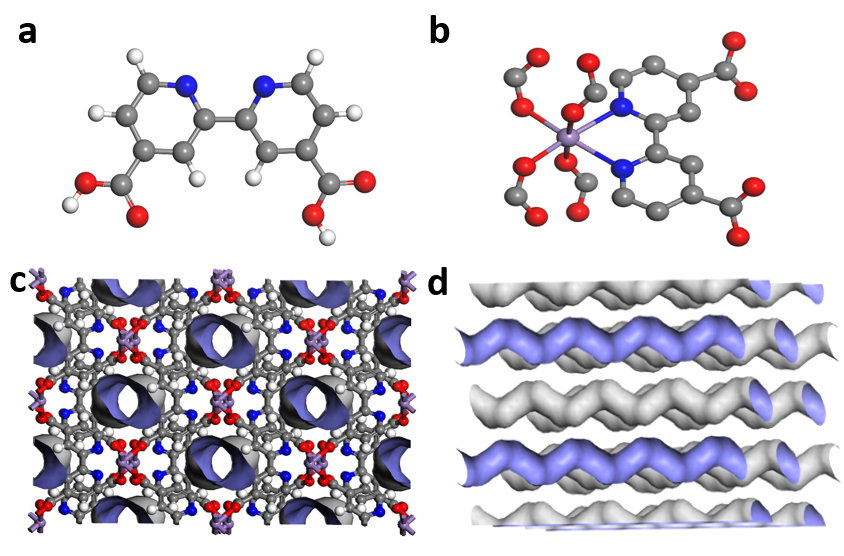
Efficient and economical separation of 1,3-butadiene (C4H6) from C4 hydrocarbons is imperative yet challenging in industrial separation processes. Herein, a guest-induced flexible Mn-bpdc MOF has been employed to separate C4H6 from C4 hydrocarbons, including n-butene (n-C4H8), iso-butene (iso-C4H8), n-butane (n-C4H10) and iso-butane (iso-C4H10). Significantly, C4H6 can instantaneously induce gate-opening of Mn-bpdc MOF at 0.13 bar and 298 K, thus significant amounts of C4H6 can be adsorbed, while other C4 hydrocarbons cannot induce the gate-opening even at 1 bar. The uptake selectivities of Mn-bpdc MOF for C4H6/n-C4H8 and C4H6/iso-C4H8 are up to 40.0 and 45.0 at 298 K and 1 bar, respectively, both surpassing all the reported adsorbents. In addition, breakthrough experiments verified that C4H6/n-C4H8, C4H6/iso-C4H8, C4H6/n-C4H10 and C4H6/iso-C4H10 mixture can be efficiently separated. More importantly, Mn-bpdc possesses excellent water stability and outstanding regeneration ability for C4H6 separation, making it a new benchmark for C4H6 purification.