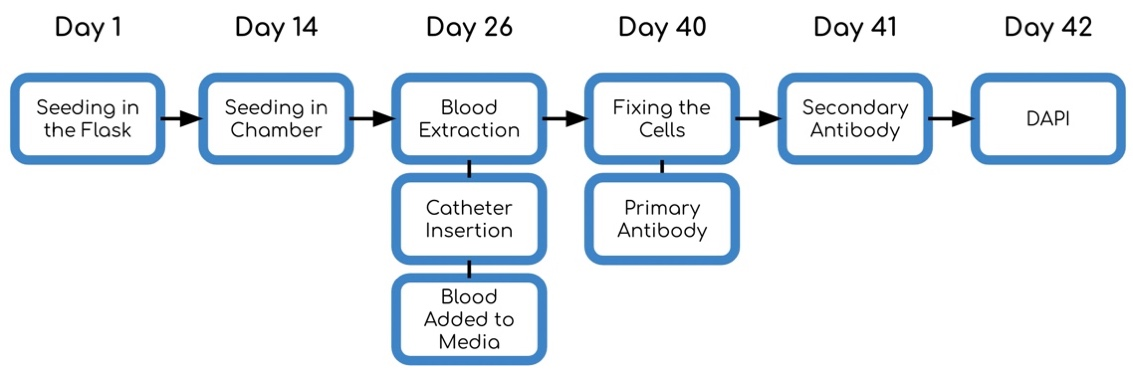
Otitis Media (OM) is the most common reason for U.S. children to receive prescribed oral antibiotics, leading to potential to cause antibiotic resistance. To minimize oral antibiotic usage, we developed polyvinylpyrrolidone-coated silver nanoparticles (AgNPs-PVP), which completely eradicated common OM pathogens, i.e., Streptococcus pneumoniae and non-typeable Haemophilus influenzae (NTHi) at 1.04µg/mL and 2.13µg/mL. The greater antimicrobial efficacy against S. pneumoniae was a result of the H2O2-producing ability of S. pneumoniae and the known synergistic interactions between H2O2 and AgNPs. To enable the sustained local delivery of AgNPs-PVP (e.g., via injection through perforated tympanic membranes), a hydrogel formulation of 18%(w/v)P407 was developed. Reverse thermal gelation of the AgNPs-PVP-P407 hydrogel could gel rapidly upon entering the warm auditory bullae and thereby sustained release of antimicrobials. This hydrogel-based local delivery system completely eradicated OM pathogens in vitro without cytotoxicity, and thus represents a promising strategy for treating bacterial OM without relying on conventional antibiotics.